
先天性白内障12个家系基因突变分析
2022-11-18 09:51:30白周现邵敬芝刘莉娜孔祥东
中华实验眼科杂志 2022年10期
白周现 邵敬芝 刘莉娜 孔祥东
1郑州大学第一附属医院遗传与产前诊断中心,郑州 450000;2郑州大学第一附属医院眼科,郑州 450000
先天性白内障是主要的儿童致盲眼病之一,婴幼儿出生时或1岁前可出现部分或全部晶状体混浊,严重影响患儿视力发育。先天性白内障可分为仅有晶状体混浊的单纯白内障和伴有其他眼部及全身其他系统异常的综合征;单纯白内障也可伴或不伴其他眼部异常,如小角膜、小眼球和虹膜缺损等[1]。先天性白内障与遗传关系密切,遗传性白内障常见的遗传方式为常染色体显性遗传,少数为常染色体隐性遗传及X染色体连锁隐性遗传[2]。先天性白内障大多数是单纯型,通常双眼受累,单眼受累较少见[3]。目前已报道有数十个基因的相关变异可导致先天性白内障,包括晶状体蛋白基因、膜蛋白基因、调节眼球发育的基因、热休克蛋白等,其中晶状体蛋白基因数量最多[4]。然而,目前已知的致病性基因变异并不能完全解释所有的遗传相关先天性白内障,高通量测序技术在遗传性眼病临床检测中的实践为发现更多的致病性变异提供了有效手段。本研究拟通过分析先天性白内障患者的临床表现和致病变异,为先天性白内障患者产前诊断提供理论依据。
1 资料与方法
1.1 一般资料
采用家系调查研究,纳入2018年1月至2019年12月于郑州大学第一附属医院就诊的12个无血缘关系的中国汉族先天性白内障家系,收集其既往病史及相关检查。纳入本研究的27例患者均为检出致病基因的先天性白内障患者。本研究遵循《赫尔辛基宣言》,研究方案经郑州大学第一附属医院伦理委员会批准(批文号:KS-2018-KY-36),受试者或其监护人均对本研究知情并自愿签署知情同意书。
1.2 方法
1.2.1捕获测序及变异位点分析 采集患者和家系成员外周血各4 ml。外周血基因组DNA使用Blood DNA Midi Kit D3494试剂盒(美国Omega Bio-Tek公司),通过epMotion全自动核酸提取纯化工作站(德国Eppendorf公司)抽提。利用Agilent SureSelect外显子靶向序列富集系统进行序列捕获(含153个白内障致病基因,北京贝瑞和康公司设计),采用Illumina NextSeq 500(美国Illumina公司)测序。目标序列测序深度平均为100×。变异注释选择GRCh37版人类基因组参考序列,筛选分析得到候选致病变异位点。入选本研究候选致病变异的条件为患者疾病表型、家族史、相关致病基因的遗传方式均符合先天性白内障。
候选变异致病性分析通过人类基因变异数据库(human gene mutation database,HGMD)、单核苷酸多态性数据库(database of single nucleotide polymorphisms,dbSNP)查询目标变异收录情况。变异的人群频率检索基因组整合数据库gnomAD。候选变异报道情况和致病性研究通过PubMed数据库查询。由GERP++软件工具分析变异位点的氨基酸序列保守性;采用SIFT、PolyPhen_2、MutationTaster蛋白功能分析软件预测错义变异的影响,判断其致病性,其中SIFT预测值范围为(0,1),值越小越有害;PolyPhen_2和MutationTaster预测值范围为(0,1),值越大越有害。
1.2.2变异验证 参照本研究团队前期研究结果,采用实时荧光定量PCR法验证家系12致病基因外显子缺失重复[5]。其他家系的候选变异行Sanger测序法验证。采用Taq DNA聚合酶(立陶宛Fermentas公司)和特异引物(GeneTool设计)对受试者基因组DNA目标序列进行扩增,其中3个新突变家系的引物设计见表1。PCR扩增条件:95 ℃预变性5 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸45 s,循环32次;72 ℃终延伸10 min,纯化(乙醇/醋酸钠法按BigDyeV3.1操作)DNA片段产物变性后,通过3130xl型测序仪(美国ABI公司)用dGTP BigDye®Terminator Sequencing Kit(美国ABI公司)进行检测,反应条件为96 ℃ 1 min;96 ℃ 10 s,50 ℃ 5 s,60 ℃ 4 min,循环25次。
2 结果
2.1 12个家系患者临床特点
12个家系家系图见图1,其主要临床表现见表2,其中单纯白内障共9个家系(家系1~9),先天性白内障伴无虹膜症1个家系(家系10),综合征型白内障2个家系(家系11和12)。除家系11患者Ⅱ1为单眼白内障外,其他患者均双眼患病。家系6患者Ⅱ1和家系10的各个患者除白内障外,还表现出眼球震颤,家系7的每例患者均表现出小眼球症(图2)。

表1 3个新变异的Sanger测序验证引物Table 1 Primers for identification of novel variations by Sanger sequencing家系基因位点正向引物序列(5'-3')反向引物序列(5'-3')产物长度(bp)5CRYGDc.422GAGGACTACAGAGGCCAGATGATATGCCAGGAACACACAGAAAATATT3057CRYBB2c.434CGTAGTGGGTGCACTGGGCAGAGGTCAGCAGAGCACACT40510ELP4c.886GGGGAGACGATATTTGCTGTGCGCAAATGGCCAGAAAGCACCT264
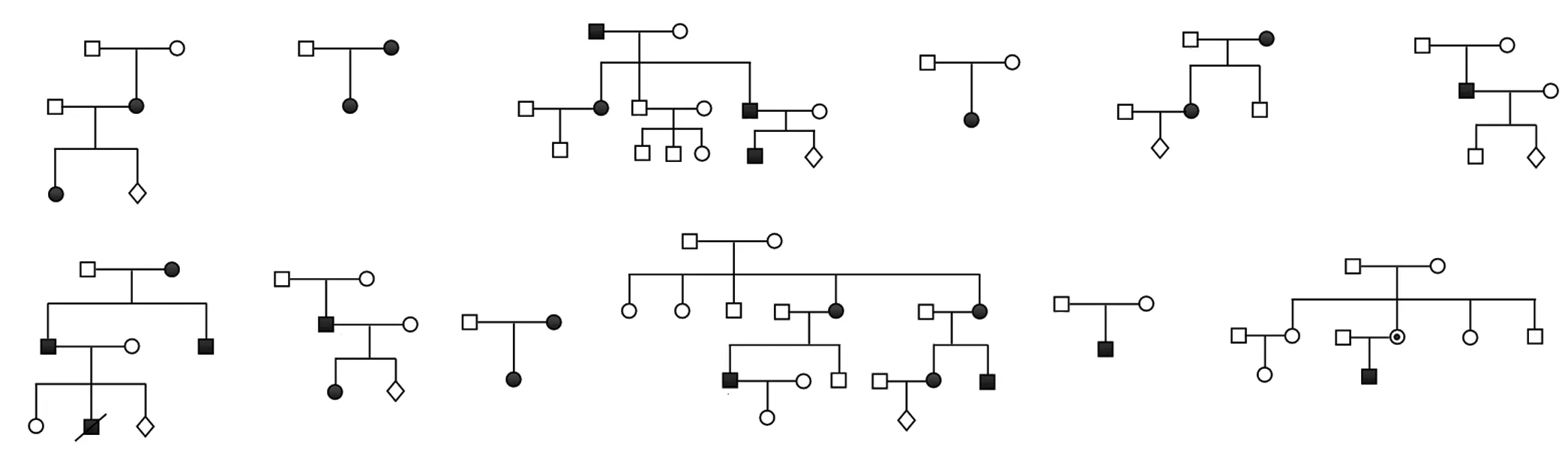
图1 12个先天性白内障家系家系图 □:正常男性;○:正常女性;■:男性患者;●:女性患者;:性别未知胎儿;:女性携带者;/:已故;:先证者Figure 1 Pedigrees of 12 families with congenital cataract □:normal male;○:normal female;■:male patient;●:female patient;:fetus of unknown sex;:female carrier;/:deceased;:proband

表2 12个先天性白内障家系患者临床表现Table 2 Clinical manifestations of patients with congenital cataract in 12 collected pedigrees家系患者例数患者主要临床表现12患者Ⅱ2,女,32岁,双眼先天性中央核性白内障;患者Ⅲ1,女,4岁,双眼先天性白内障22患者Ⅱ 1,女,19岁,出生3 d发现双眼白内障,4月龄时行白内障手术,术后视力基本正常;患者Ⅰ2,女,44岁,双眼先天性白内障(晶状体外周点状混浊)、视力障碍34患者Ⅲ5,男,7岁,1岁发现双眼先天性白内障,行白内障摘出术;患者Ⅰ1、Ⅱ2和Ⅱ5双眼先天性白内障41患者Ⅱ1,女,29岁,双眼先天性白内障52患者Ⅱ2,女,26岁,双眼先天性白内障;患者Ⅰ2双眼先天性白内障61患者Ⅱ1,男,42岁,双眼先天性白内障、眼球震颤、斜视、弱视74患者Ⅱ1,男,37岁,双眼先天性白内障、双眼小眼球;患者Ⅰ2和Ⅱ3双眼先天性白内障、双眼小眼球;患者Ⅲ2,胎儿期经基因产前诊断为先天性白内障患者即引产82患者Ⅱ1,男,28岁,双眼先天性白内障;患者Ⅲ1双眼先天性白内障92患者Ⅱ1,女,26岁,双眼先天性白内障;患者Ⅰ2双眼先天性白内障105患者Ⅲ5,女,22岁,双眼先天性白内障、眼球钟摆样震颤、虹膜缺损;患者Ⅱ5、Ⅱ7、Ⅲ1和Ⅲ6双眼先天性白内障、虹膜缺损、眼球震颤111患者Ⅱ1,男,3岁,右眼先天性白内障,左眼视盘发育异常,肾脏发育异常121患者Ⅲ2,男,2岁,双眼先天性白内障,蛋白尿,脑发育迟缓
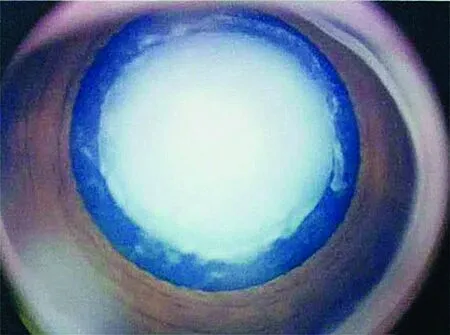
图2 家系11患者Ⅱ1右眼白内障照片Figure 2 Right eye with cataract of patient Ⅱ1 in family 11
2.2 二代测序筛查
经Illumina NextSeq 500测序平台DNA靶向测序,12个家系患者分别检测到与其表型高度相关的基因变异(表3)。
2.3 候选变异验证
各个基因变异验证结果均与二代测序一致,3个新变异Sanger测序验证结果见图3。12个家系各基因变异型和疾病表型全部共分离。12个家系受试者的相关变异携带情况见图1。

表3 先天性白内障12个家系基因变异概况Table 3 Genetic variations of 12 families with congenital cataract included家系基因核酸改变氨基酸改变合子状态人群频率致病性预测遗传方式疾病[OMIM number]新变异(文献)1CRYBA4c.277T>Cp.S93PHet-+++AD白内障23型[610425]否[6]2MIPc.97C>Tp.R33CHet-+++AD白内障15型[615274]否[7]3CRYBB2c.563G>Ap.R188HHet-+++AD白内障3型[601547]否[8]4CRYBB2c.436G>Cp.V146LHet-+-+AD白内障3型[601547]否[9]5CRYGDc.422delGp.G141Dfs∗6Het-///AD白内障4型[115700]是6GJA8c.593G>Ap.R198QHet-+++AD白内障1型[116200]否[10]7CRYBB2c.434G>Cp.R145PHet-+++AD白内障3型[601547]是8CRYGCc.470G>Ap.W157XHet-+++AD白内障2型[604307]否[11]9CRYGDc.70C>Ap.P24THet----AD白内障4型[115700]否[12]10ELP4c.886C>Ap.L296IHet-+++AD无虹膜2型[617141]是11PAX2c.70dupGp.V26Gfs∗28Het0.000 2///ADRCS[120330]否[13]12OCRLE5-E16dup-Hemi-///XRLowe综合征[309000]否[5] 注:OMIM:人类在线孟德尔遗传数据库;Het:杂合变异;Hemi:半合子变异;AD:常染色体显性遗传;XR:X染色体连锁隐性遗传;RCS:肾视神经乳头缺损综合征 致病性分别由SIFT、PolyPhen_2、MutationTaster预测,+表示预测有害或致病,-表示预测良性,/表示无结果 Note:OMIM:Online Mendelian Inheritance in Man;Het:heterozygous variation;Hemi:hemizygous variation;AD:autosomal dominant inheritance;XR:X-linked recessive inheritance;RCS:renal coloboma syndrome The pathogenicity was predicted by SIFT,PolyPhen_ 2 and MutationTaster,respectively,and+indi-cated damaging,- for benign,/ for NA
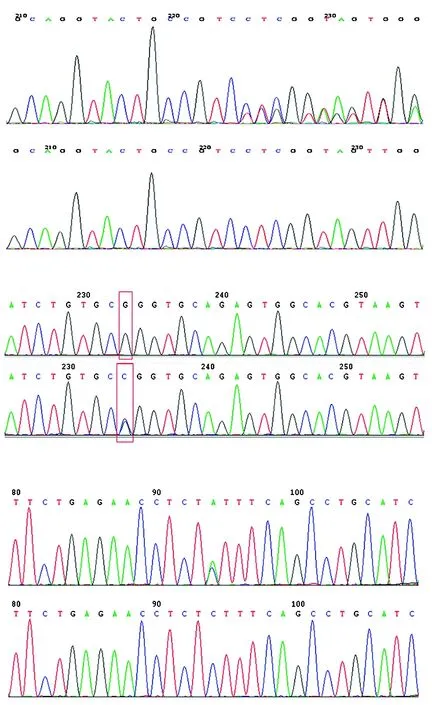
图3 3个新变异Sanger测序验证结果 A:家系5 CRYGD基因c.422delG突变型与野生型 B:家系7 CRYBB2基因c.434G>C突变型与野生型 C:家系10 ELP4基因c.886C>A突变型与野生型Figure 3 Sanger sequencing analysis for validation of 3 new variations A:Mutant and wild-type CRYGD c.422delG in family 5 B:Mutant and wild-type CRYBB2 c.434G>C in family 7 C:Mutant and wild-type ELP4 c.886C>A in family 10
2.4 变异致病性分析
12个先天性白内障家系共检出12个不同候选致病变异(表3),其中ELP4基因c.886C>A、CRYBB2基因c.434G>C、CRYGD基因c.422delG在PubMed数据库和HGMD专业版均未见报道,为新发现的变异位点。CRYBA4基因c.277T>C和OCRL基因E5-E16dup为本课题组已报道变异[5-6]。CRYGD基因c.422delG为移码变异,依据《遗传变异分类标准与指南》和2015年版《ACMG Standards and Guidelines》判断其致病性非常强,等级为PVS1,为致病性变异,主要表现为翻译过程提前终止而产生了截短的无功能产物蛋白。
ELP4基因c.886C>A和CRYBB2基因c.434G>C位点在dbSNP数据库中无记录编号,不属于多态性位点,人群发生频率极低。gnomAD数据库查询该变异在人群中发生频率未记录。蛋白功能分析软件对这2个变异位点进行预测,SIFT预测值为0,提示为有害变异,PolyPhen_2和MutationTaster预测值均为1,提示为有害和致病变异。ELP4基因c.886C>A变异导致第296位氨基酸由非极性二级结构趋向α螺旋的脂肪族亮氨酸替换为非极性二级结构趋向β折叠的异亮氨酸,CRYBB2基因c.434G>C变异导致第145位氨基酸由极性带正电无二级结构趋向的脂肪族精氨酸替换为非极性二级结构趋向β转角的杂环脯氨酸。CRYBB2基因c.436G>C(p.V146L)是与c.434G>C位置紧临的已知致病性错义变异[9],c.434G>C有害的可能性大。ELP4基因c.886C>A(p.L296I)和CRYBB2基因c.434G>C(p.R145P)位点氨基酸序列在物种间高度保守(图4),变异有害的可能性大。

图4 ELP4基因296位和CRYBB2基因145位氨基酸序列在物种间的保守性 2个位点均高度保守Figure 4 Conservation of amino acid sequences at position 296 of ELP4 and position 145 of CRYBB2 among species Both loci were highly conserved
3 讨论
本研究12个先天性白内障家系包括9个单纯白内障家系、1个先天性白内障伴无虹膜症家系和2个综合征型白内障家系。这些家系中仅RCS为单眼白内障,其余均为双眼发病;依据孟德尔遗传定律,仅眼脑肾综合征为X染色体连锁隐性遗传,其余均为常染色体显性遗传。针对晶状体致病基因靶向捕获测序进行的分析发现,12个家系致病基因变异可分为3个组,即晶状体蛋白类变异(家系1、3、4、5、7、8、9)、膜转运蛋白类变异(家系2、6、12)和转录因子复合体类变异(家系10、11)。本研究通过临床表型结合遗传学分析确诊了12个先天性白内障家系的致病基因及变异位点。
晶状体蛋白占人晶状体湿重的30%~40%,占晶状体干重的90%。α、β、γ晶状体球蛋白均为遗传性突变的靶蛋白,这些蛋白的基因变异可导致蛋白质结构异常和晶状体混浊[14],从而引起先天性白内障。本研究检出的晶状体球蛋白类变异基因包括CRYBA4、CRYBB2、CRYGC和CRYGD。CRYBA4基因编码β晶状体球蛋白A4亚基,β晶状体球蛋白是一种复杂的蛋白聚合体,通常同种亚基结合为同二聚体或不同亚基聚合为异二聚体。CRYBA4为β晶状体球蛋白酸性基团成员,是与β-B1、β-B2和β-B3组成的基因簇的一部分[15]。CRYBB2基因编码β晶状体球蛋白B2亚基。CRYGC为γ晶状体球蛋白家族的成员之一,据报道其变异与核性粉状白内障和板层白内障有关[12,16]。CRYGD基因变异可以出现更多的表型变异。γ晶状体球蛋白是一组由高度对称的单体蛋白组成的同源基因,通常缺乏连接肽和末端延伸。γ晶状体球蛋白家族包括4个基因(γ-A、γ-B、γ-C、γ-D)和3个假基因(γ-E、γ-F、γ-G),在基因组序列中以基因簇的形式排列。晶状体的透明特性是组织结构高度有序的结果,这些结构的维持依赖于水合作用和单个细胞容积的控制。而各种相关膜转运蛋白就承担着这样的任务,其基因变异可导致先天性白内障[14]。本研究检出的膜转运蛋白类变异基因包括MIP、GJA8和OCRL。MIP基因编码晶状体纤维的主要内在蛋白AQP0,是成熟晶状体内最多的连接膜蛋白,作为水通道蛋白涉及快速的水跨膜运动。GJA8基因编码缝隙连接蛋白α-8,作为缝隙连接蛋白的一部分,连接素Cx50是晶状体纤维细胞生长和成熟必需的。OCRL基因编码肌醇多磷酸盐5-磷酸酶,涉及调节膜转运过程,也定位于多个亚细胞膜。研究表明,OCRL基因产物定位于视网膜色素上皮细胞、成纤维细胞和肾小管细胞的初级纤毛,OCRL变异导致纤毛缩短、纤毛功能障碍[17],从而使眼、肾脏出现相应症状。初级纤毛信号通路也介导眼压感觉[18]。最新的RNA测序研究在OCRL变异导致的眼脑肾综合征中发现了与眼部白内障相关的差异表达基因[19]。晶状体的发育是由多种基因在时间和空间上相互作用共同协调完成的,转录因子基因变异会导致一定程度的眼前节,如角膜和晶状体发育异常[20]。本研究检出的转录因子复合体类变异基因有ELP4和PAX2。ELP4基因编码延伸子乙酰转移酶复合物亚基4,在转录延伸期间直接与RNA聚合酶Ⅱ接合。ELP4基因对PAX6的表达有下游转录调控作用[21],PAX6为已知的无虹膜症致病基因,可导致无虹膜及白内障表型。ELP4基因变异可间接通过调控PAX6影响白内障表型。PAX2基因编码配对盒2,该转录因子基因家族的核心特征是保守的DNA结合配对盒结构域。
靶向测序能够辅助先天性白内障遗传病因的确诊及亚型鉴别。Astiazarn等[22]对11个墨西哥先天性白内障家系进行了靶向捕获测序研究,Zhai等[23]对27个中国先天性白内障家系进行了外显子组靶向捕获测序研究,其结果同样涉及本研究结果所属的3类基因,分别达到了55%(6/11,墨西哥人)和63%(17/27,中国人)的分子诊断率。本研究分子诊断率约为71%(12/17),这可能与根据临床表型纳入的白内障受试者和靶向捕获基因的优化有关。本研究12个家系患者的临床表现显示了异质性,家系11、12为伴有白内障的综合征患者,家系7白内障3型患者均双眼球缩小,而家系3、4患者无此症状。家系10无虹膜2型患者均虹膜缺损轻微,以白内障表现为主。一些白内障患者还表现出眼球震颤。本研究结果表明,不同基因型的先天性白内障表现有差异,即使相同基因型的先天性白内障表现也可不同。
本研究汇总分析了12个不同家系先天性白内障患者的临床表现和致病基因变异,报道了导致先天性白内障的3个新变异并分析了变异致病性。本研究结果拓展了常染色体显性遗传及X染色体连锁隐性遗传的先天性白内障致病基因变异谱,丰富了先天性白内障相关表型谱,有助于进一步理解该病的分子机制,为先天性白内障的确诊、遗传咨询和产前诊断提供了理论依据。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明白周现:参与选题、酝酿和设计试验、起草文章;邵敬芝:实施研究、采集数据;刘莉娜:分析/解释数据;孔祥东:对文章知识性内容的审阅和智力性内容的修改及定稿
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21 02:14:52
中国实用医药(2021年7期)2021-04-16 11:21:38
心肺血管病杂志(2020年5期)2021-01-14 00:43:30
中医眼耳鼻喉杂志(2019年2期)2019-04-13 05:23:52
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
中外医疗(2015年5期)2015-08-29 01:54:42
中国当代医药(2015年30期)2015-03-01 02:08:19
西南国防医药(2015年11期)2015-02-28 19:38:55
