
Rosai-Dorfman病的影像学特征
2022-10-15 04:00黄小姬周莉刘斐杜丽娜
中国医学影像学杂志 2022年9期
黄小姬,周莉,刘斐,杜丽娜
南昌大学第一附属医院放射科,江西 南昌 330000;*通信作者 周莉 813136252@qq.com
窦组织细胞增生伴巨大淋巴结病(Rosai-Dorfman disease,RDD)是一种特发性组织细胞增生性疾病,最早于1969年被描述[1],由于本病罕见,其发病机制及诸多特征尚未阐明。目前,关于RDD的研究多数为个案报道,大样本研究较为少见,本研究回顾性分析12例经病理证实为RDD的影像学特点,并复习相关文献,以增加对本病的了解,为本病的诊疗提供新的见解。
1 资料与方法
1.1 研究对象 收集南昌大学第一附属医院2017年11月—2021年9月收治的12例RDD,其中男8例,女4例,年龄17~65岁,平均(52±13)岁,临床表现主要为无痛性进行性肿大的软组织包块、头痛头晕、肢体纳入标准:行CT或MRI检查,临床及病理资料均完整,符合RDD诊断标准。本研究经本院伦理委员会批准[〔2022〕医研伦快审第(3-023)号]。
1.2 检查方法 5例行CT平扫, 3例行CT平扫+增强扫描,采用Siemens CT 64层128排扫描(管电压120 kV,管电流150~250 mAs,层厚1 mm),按相应部位标准划分扫描范围。经静脉注射碘海醇(1.5 ml/kg)行增强扫描。
8例行MRI平扫+增强扫描,其中4例同时行CT检查,采用Siemens 3T MRI扫描,扫描序列为常规SE T1WI(TR 250 ms,TE 2.46 ms,层厚5 mm)、TSE T2WI(TR 4 000 ms,TE 113 ms,层厚5 mm)、FLAIR序列(TR 8 000 ms,TE 2 722 ms,层厚5 mm)及扩散加权成像(DWI)扫描,b值为0、800~1 000 s/mm2,经静脉注入钆喷酸葡胺(0.2 mmol/kg)后获取轴向、矢状及冠状T1WI增强扫描(矩阵256×256,视野24×24,层厚5 mm,层间距1 mm)。另1例右侧尺骨RDD采用Philips DR摄片机,管电压57 kV,管电流10 mAs。
1.3 图像分析 由2位影像科主任医师采用盲法观察病变部位、形态、大小、边缘、信号/密度特点、强化方式及邻近结构改变等,意见出现分歧时经过讨论达成一致。
1.4 病理及免疫组化 所有标本经甲醛处理,取材,切片后于光镜下观察;免疫组化染色主要标志物包括CD68、S-100蛋白、CD1a、EMA、Ki-67、ALK、Langerin、IgG及IgG4等。
2 结果
2.1 一般资料与影像学表现 12例RDD患者的临床表现与影像学表现见表1、图1~5。

表1 12例RDD患者的临床资料与影像学表现
病变部位 :纯结外型9例,纯结内型2例,混合型1例。9例纯结外型中,中枢神经系统6例、鼻腔及鼻旁窦2例、骨肌系统1例。
临床表现:3例出现头痛,其中1例合并癫痫(病例4);2例出现肢体麻木,1例出现视物模糊,1例同时累及左侧大腿皮下软组织及右侧尺骨分别表现为大腿包块及右前臂疼痛,2例出现鼻塞,1例表现为鼻部肿物,3例表现为颈部或颌下多发肿大淋巴结。
影像表现:淋巴结型:3例累及淋巴结的病灶CT均表现为等-略高密度,其中1例淋巴结T2WI伴有低信号纤维分隔(图1A)。骨肌系统:病例7左侧大腿病灶表现为长T1WI长T2WI信号(图2A),右侧尺骨病灶于MRI表现为长T1WI长T2WI信号(图2B),CT表现为溶骨性低密度区,呈磨玻璃改变(图2C);中枢神经系统:2例行CT扫描的患者均呈稍高密度肿块,5例行MRI扫描的患者T1WI呈低-等信号(图3A),4例T2WI呈低-等信号(图4A),另1例患者T2WI呈高信号。头颈五官系统:3例鼻腔及鼻旁窦RDD患者CT均表现为等-略高密度,均累及鼻翼,病灶趋向结节样改变(图5A),其中1例显示骨质吸收破坏(图5B)。所有已行增强扫描的病灶均强化明显(图3B、4B)。

图2 女,RDD。17岁时行MRI见T2WI抑脂序列左侧大腿病灶呈高信号(箭,A);19岁时行MRI见右侧尺骨骨髓腔内多发条片状T2抑脂高信号影,局部累及骨皮质,强化明显(箭,B),CT示右侧尺骨髓腔内密度不均,呈磨玻璃样改变,骨质连续性欠佳,局部见溶骨性骨质破坏区(箭,C)
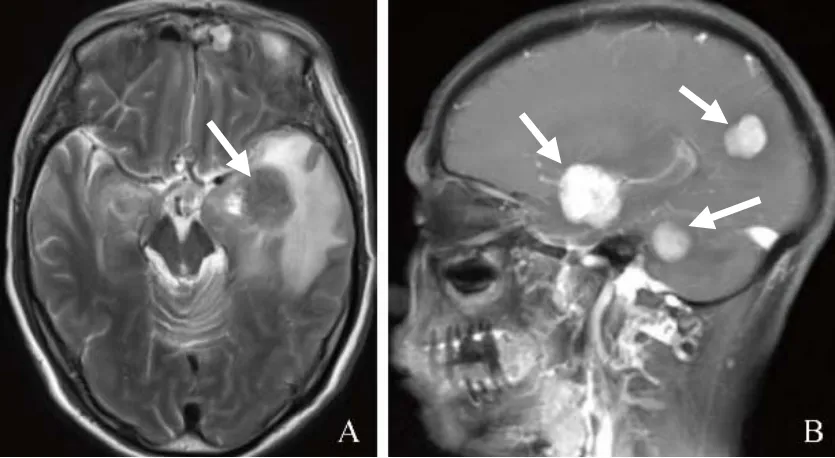
图4 男,55岁,RDD。A:双侧大脑半球、左侧小脑半球多发结节状肿块影,T2WI呈低信号(箭);B:增强扫描明显强化(箭)

图5 女,61岁,RDD。A:双侧鼻腔、鼻旁窦腔及鼻翼区不规则软组织肿块,趋向结节样改变(箭);B:骨窗示邻近骨质吸收破坏及鼻甲受侵(箭)
2.2 病理结果 12例RDD均由手术病理证实,大体上主要为灰白色或灰褐色组织,质韧。镜下主要见淡染区的组织细胞及深染区的淋巴细胞或浆细胞交替分布,可伴有间质胶原纤维组织增生及中性粒细胞浸润,局部可见“淋巴细胞吞噬作用”或“伸入运动”(图1B、图3C),累及淋巴结者淋巴窦扩张,部分淋巴结结构欠清。
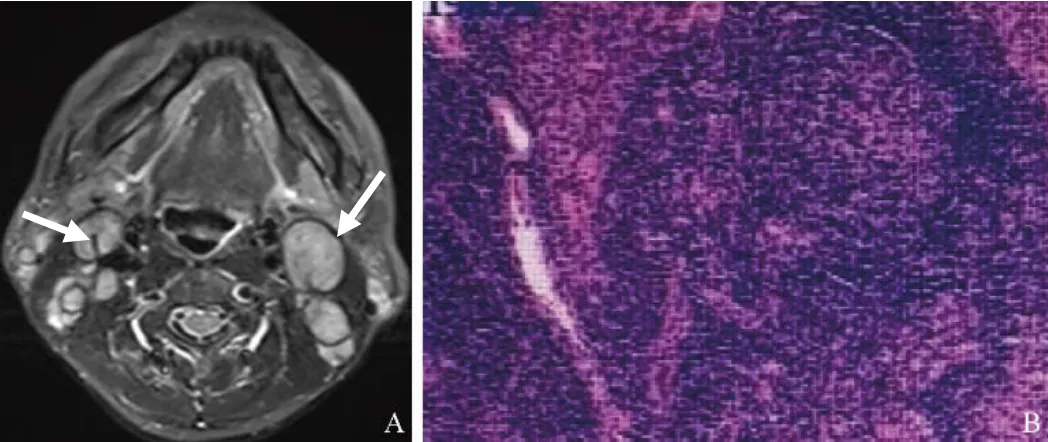
图1 男,42岁,RDD。A:MRI见双侧颈部多发肿大淋巴结(箭),部分淋巴结T2WI上见条状低信号分隔;B:淋巴结病理活检镜下见淋巴窦扩张,其间见大量组织细胞片状分布,局部见“伸入现象”(HE,×200)
2.3 免疫组化结果 12例RDD患者中,S-100和CD68均为(+)(图3D),CD1a均为(-),EMA(-)8例,Langerin(-)10例,ALK(-)10例,伴有IgG4/IgG(+)浆细胞增多8例,Ki-67增殖指数<10%。
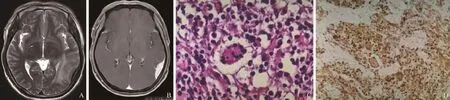
图3 男,44岁,RDD。A:T2WI示左侧颞枕部颅骨内板下梭形稍低信号影,病灶与颅板宽基底相连(箭);B:增强扫描强化明显,可见“脑膜尾征”,边缘见“毛刺征”(箭);C:病理活检镜下见空泡状组织细胞增生,局部见“噬淋巴现象”(HE,×400);D:免疫组化见组织细胞CD68免疫反应阳性(EnVision法,×100)
3 讨论
3.1 RDD的临床表现 关于RDD的起源尚不明了,可能与病毒感染、免疫调节紊乱、基因突变以及信号传导通路相关[2],也有学者提出RDD可能与IgG4之间存在关联[3],然而,目前这些假说均未得到证实。本研究发现RDD主要发生于中青年人群,男性更多见,与Zhu等[1]的报道一致。
RDD可分为结内型、结外型及混合型。结内型常表现为无痛性、双侧颈部大量淋巴结肿大,颌下、颏下、腋窝、纵隔、腹股沟及腮腺区淋巴结亦可累及,但腹膜后淋巴结病并不常见[4]。本组2例为纯结内型RDD,均表现为双侧颈部或颌下多发肿大淋巴结。
结外型病变约占所有RDD的43%,可累及皮肤、鼻腔、骨肌、胃肠道、泌尿生殖系统及中枢神经系统等[5]。①骨肌系统:结外型病变多累及皮肤及软组织,通常表现为界限分明的丘疹,可触及肿块[6];骨骼RDD罕见,常累及中轴骨及长骨,包括胫骨、股骨、肱骨、锁骨以及颅骨等,多表现为疼痛、肿胀等[7]。本组1例先累及左侧大腿,2年后累及右侧尺骨,此前未见尺骨RDD病变报道,查体右前臂可触及肿块,质地硬,压痛。②中枢神经系统:<5%的病例累及中枢神经系统[8],颅内RDD症状不典型,可表现为头痛、肢体乏力、癫痫发作、颅神经功能障碍和视觉障碍等[9]。本组6例颅内RDD中,3例表现为头痛,2例表现为肢体乏力,1例出现视物模糊。③头颈五官系统:鼻腔或鼻旁窦RDD常出现鼻塞、鼻出血等症状[10]。本组中1例反复鼻塞1年余,另1例发现鼻翼处肿物8个月余。本组病例显示鼻腔RDD病变较常累及鼻翼,查体可见鼻翼或鼻道内新生物、鼻中隔增厚及鼻甲肥厚。④其他系统:约2%的RDD累及呼吸系统,通常伴有淋巴结病变,包括肺间质性疾病、肺结节、气管支气管疾病等[11-12];<1%的病例累及消化系统,主要累及胃肠道、胰腺、肝脏等[13]。
混合型:本组1例既累及双侧颈部、颌下及腮腺区淋巴结,又累及双侧鼻腔,为混合型。
本研究中,纯结外型RDD较为常见(9例),而纯结内型RDD发生率较低(2例),混合型RDD最少(1例)。在器官受累方面,最常受累的是中枢神经系统(6例),鼻腔和鼻旁窦(3例)也易累及。这些趋势与既往报道不一致,有待日后扩大样本量深入研究。
3.2 RDD的组织病理及免疫组化特点 组织病理学及免疫组化是诊断RDD的“金标准”。RDD的病理组织学特点:在许多浆细胞和淋巴细胞背景下有大量体积较大的组织细胞增生,胞质嗜酸性,部分胞质内见淋巴细胞及浆细胞(即“淋巴细胞吞噬作用”),可伴有间质胶原纤维组织增生及中性粒细胞浸润,累及淋巴结者可见淋巴窦扩张,部分淋巴结结构破坏,显示不清[4]。免疫组化特点:RDD比较有特征性意义的表现是对S-100蛋白和CD68呈免疫反应性,而对CD1a免疫染色缺乏免疫反应。本组病例与上述组织病理及免疫组化表现相符。
3.3 影像表现
3.3.1 结内型 多表现为双侧颈部多发淋巴结肿大,常伴有颌下、颏下及腮腺区淋巴结肿大,淋巴结边界尚清晰,很少融合,CT一般呈等/稍低实性密度,MRI一般呈等T1稍长T2信号,本研究发现部分淋巴结内T2WI常伴有较多低信号纤维分隔,未见坏死囊变,呈欠均匀明显强化。该影像特征可能对结内型RDD具有一定的提示作用,但仍需进一步研究。
3.3.2 结外型 累及不同部位时具有对应的影像特征,主要表现为:①累及皮肤或皮下软组织常表现为长T1长T2信号皮下软组织结节或肿块影,T2WI抑脂及DWI高信号,边界多清晰。②累及骨骼主要表现为溶骨性病变,主要集中于骨髓腔内,可呈虫蚀状改变,很少伴有周围软组织肿块及骨皮质破坏,骨膜反应及骨质硬化很少发生,MRI对于病变更为敏感,可以早期发现病变[14-18]。本组1例患者先后累及左侧大腿皮肤及右侧尺骨,其中左侧大腿皮下病灶表现与上述描述大致相符,该患者右侧尺骨内可见多发溶骨性骨质破坏区,病灶主要局限于骨髓腔内,部分沿着骨髓腔蔓延并局部突破骨皮质致骨质破坏。③颅内RDD多数累及硬脑膜,脑实质及脑室内病变罕见[1]。本组6例颅内RDD病例中,1例位于鞍区,2例累及硬脑膜,另3例累及脑实质(其中1例表现为脑实质内多发病灶)。颅内RDD在CT上主要表现为稍高密度肿块,无明显钙化[19],T1WI多呈等信号,T2WI多呈低-等信号,增强扫描硬脑膜型常可见“硬膜尾征”,术前多误诊为脑膜瘤[20],但颅内RDD多见于中年男性,而脑膜瘤多见于中老年女性。另外,本研究发现硬脑膜型RDD硬脑膜累及的范围更广,病灶可沿脑沟脑裂向颅内延伸,呈“毛刺征”改变,有助于两者的鉴别。本组病例中1例脑实质型RDD T2WI呈高信号,颅内RDD病灶在T2WI呈高信号鲜有报道,结合该患者病理结果推测此高信号可能与病程或病灶内细胞种类含量相关,提示病变处于病程急性期或与该病变伴有的淋巴细胞及浆细胞浸润比较少,而以大量组织细胞增生为主,导致T2WI信号偏高。④头颈五官系统:本研究发现鼻翼是鼻腔RDD最常受累的部位,本组中1例表现为鼻翼处肿块,另2例除鼻翼处软组织肿块外同时累及鼻腔或鼻旁窦,表现为腔内不规则软组织密度影,部分肿块趋向结节样改变,常伴鼻中隔增粗及鼻甲肥厚,随着病变进展可伴有骨质的吸收破坏及鼻甲受侵,增强扫描病变亦呈明显强化,鼻腔及鼻旁窦RDD病变需与Wegener肉芽肿相鉴别,两者均为肉芽肿相关性病变,病灶信号及密度特点类似,Wegener肉芽肿晚期邻近骨质及中线结构的破坏可形成较大空腔,而鼻腔及鼻旁窦RDD病变形成大空腔未见明确报道。
总之,RDD是一种罕见疾病,机制尚未明了,可累及全身多部位,主要表现为淋巴结病或结外型病变,临床表现不典型,本研究总结并提出一些对RDD诊断具有提示意义的影像学征象,结合病理及免疫组化检查,有助于RDD的早期诊断与及时治疗。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
医学食疗与健康(2021年27期)2021-05-13
养生保健指南(2019年11期)2019-12-17
学习与科普(2019年15期)2019-09-10
恋爱婚姻家庭·养生版(2017年5期)2017-05-04
恋爱婚姻家庭(2017年15期)2017-03-17
家庭医药·快乐养生(2017年2期)2017-03-01
中国民族民间医药·下半月(2016年8期)2016-10-24
中国民族民间医药·下半月(2014年5期)2014-12-02
中国民族民间医药·下半月(2014年4期)2014-09-26
