
一个纤维素降解菌群的多样性分析
2022-09-14 14:01:04赵晶晶,杨明,李宪臻,杨帆
大连工业大学学报 2022年4期
赵 晶 晶, 杨 明, 李 宪 臻, 杨 帆
( 大连工业大学 生物工程学院, 辽宁 大连 116034 )
0 引 言
纤维素类生物质能源的开发对缓解化石燃料的短缺具有重要意义[1]。由于纤维素结构高度复杂以及纤维素生物降解效率较低,导致纤维素生物转化比较困难,从而阻碍了工业上利用纤维素生产生物燃料的进程[2]。因此,目前的研究应着眼于高效降解纤维素生物系统的挖掘。
已有研究利用16S rRNA基因扩增子测序分析技术解析了牛瘤胃[3]、牛粪堆肥[4]、森林土壤[5]等不同生境纤维素群落组成和结构,但是对于同一生境中纤维素降解过程中群落的动态变化分析尚未见报道。本实验拟利用16S rRNA基因扩增子测序分析技术对一个纤维素降解菌群(CDM)以不同纤维素代谢物为底物时物种组成及结构进行动态分析,旨在进一步阐明纤维素降解过程中群落微生物之间的动态协同作用关系,以期获得高效纤维素降解生物系统。
1 材料与方法
1.1 试剂与仪器
固体无机盐培养基:NaNO3、MgSO4·7H2O、KCl 0.5 g/L,K2HPO41 g/L,FeSO4·7H2O 0.01 g/L,琼脂20 g/L,pH 7.0。
葡萄糖/纤维二糖平板培养基:固体无机盐培养基中添加10 g/L葡萄糖或纤维二糖。滤纸平板培养基:将Whatman滤纸(直径约8.5 cm)干热灭菌后,平铺在固体无机盐培养基上。
梯度PCR仪,德国艾本德股份公司;生物分析仪,美国安捷伦科技公司;测序仪,美国因美纳公司;DYY-11型电泳仪,北京市六一仪器厂;DNA提取试剂盒,美国MoBio公司;胶回收试剂盒,美国康宁公司;测序文库构建试剂盒,美国因美纳公司;NanoDrop超微量分光光度计,美国赛默飞世尔科技公司。
1.2 方 法
1.2.1 以不同纤维素代谢产物为碳源的菌群CDM培养
选用的群落为实验室前期构建的一个稳定的纤维素降解菌群(CDM)[6]。将CDM分别接种于滤纸、纤维二糖和葡萄糖3种不同碳源的固体培养基,在30 ℃恒温培养箱中对CDM进行静置培养,以模拟纤维素降解过程:在滤纸培养基上培养3 d后有明显的菌落出现,培养8 d后滤纸开始降解,采集这两个时间点的菌落样本,分别命名为F1A和F2A,代表纤维素降解前期和中期;在纤维二糖培养基培养2 d和葡萄糖培养基上培养2 d 的菌落样本分别命名为X1A和G1A,代表纤维素降解后期和末期。
1.2.2 菌落样本的DNA提取
收集1 g附着在各个培养基上的纤维素分解菌群CDM样本,使用DNA提取试剂盒提取总基因组DNA。Nanodrop和凝胶电泳用于确定DNA质量,在构建文库之前将高质量的DNA储存在-20 ℃。
1.2.3 16S rRNA的扩增、测序与建库
以宏基因组DNA为模板,以16S rRNA V6可变区的特异性通用引物967F(5′ CAACGCGAAGAA- CCTTACC 3′)和1 046R(5′ CGACAGCCATGCA- CACCT 3′)利用LA Taq酶扩增V6可变区片段。PCR扩增程序:95 ℃变性5 min;95 ℃变性30 s;58 ℃ 复性30 s,72 ℃延伸 30 s,30个循环;72 ℃延伸10 min。PCR扩增产物使用胶回收试剂盒纯化回收,再利用NanoDrop测定PCR产物浓度。合格样品使用测序文库构建试剂盒构建测序文库,使用Agilent 2100生物分析仪对文库进行大小分布分析,并通过定量PCR进行定量。使用双末端150 bp测序策略在Illumina HiseqTM2000平台上对高质量文库进行测序。
1.2.4 生物信息学分析
16S rRNA基因扩增子测序数据生物信息学分析具体步骤:(1)测序数据的质控。原始双末端reads需要将质量值小于20的碱基占总长度20%、存在接头序列污染、不确定碱基N以及具有低复杂度(即read中不存在连续10个相同碱基)的reads过滤掉,获得高质量的clean reads;(2)标签序列的拼接和过滤。应用FLASH软件将每对高质量的双末端reads拼接成标签序列,并对这些标签序列进行过滤获得高质量的有效标签序列;(3)操作分类单元(OTU)聚类和丰度统计。应用USEARCH软件(http://www.drive5.com/usearch)OTU聚类。将OTU比对到GOLD数据库(https://gold.jgi.doe.gov)以去除PCR扩增产生的嵌合体,再将标签序列重新比对回OTU序列得到每个样品在每个OTU的丰度统计表;(4)OTU序列的物种注释。应用RDP classifier(https://sourceforge.net/projects/rdp-classifier)将OTU与Greengenes数据库(http://greengenes.lbl.gov)比对进行物种注释,并根据注释结果计算每个样品在各层次的物种信息及丰度分布情况。
1.2.5 统计分析
α多样性指数包括观察物种数、赵氏指数、艾斯指数、香农-维纳指数、辛普森指数,使用mothur 软件(https://www.mothur.org)计算,并用R软件(https://www.r-project.org)绘制稀释曲线判断测序数据量的合理性。使用β多样性来分析样本在物种结构上的差异。通过QIIME(http://qiime.org/index.html)进行基于非加权UniFrac距离的β多样性分析。
2 结果与讨论
2.1 菌群CDM的测序数据统计
采用不同纤维素代谢产物为碳源培养菌群CDM的方法来模拟纤维素降解过程,并考察该过程中CDM结构和多样性的变化。通过对4种样本F1A、F2A、X1A和G1A的16S rRNA进行高通量测序,经过拼接和过滤处理,共获得1 337 545条有效标签序列,以最低97%的序列相似度划分为相同的OTU,共获得315个细菌OTU。对4个样本进行α多样性分析,结果如表1所示。在F2A样本中CDM细菌群落数最多;F1A和X1A样本中CDM细菌群落数次之;G1A样本中CDM细菌群落数最少。
2.2 菌群CDM的α多样性比较
从表1中艾斯指数和赵氏指数两个指标可以看出,F2A的物种丰富度略高于F1A、X1A次之,G1A的物种丰富度最低,但4个样本的OTU由高到低顺序为F2A、X1A、F1A、G1A,根据赵氏指数和艾斯指数的算法推测这可能是与F1A样本中的低丰度物种较多有关[7-8]。从香农指数和辛普森指数两个指标来看,4个样本的细菌多样性由高到低顺序为F2A、F1A、X1A、G1A。
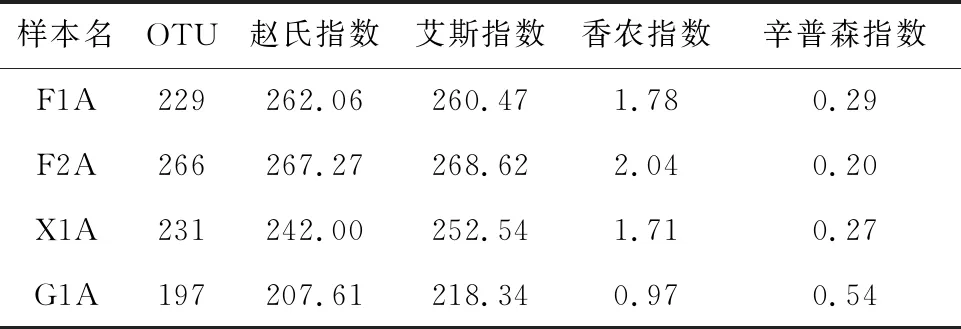
表1 菌群CDM的α多样性指数
稀疏曲线(图1(a))趋于平缓,表明4个时期的样本测序数据量合理,即各个样本得到的测序数据可以代表各个样本的物种信息。从丰度曲线可以看出物种丰度的均一性,该图中曲线的水平方向的宽度反映了物种的丰富度。从图1(b)可以看出,4个样本物种丰度均一性由高到低顺序为F2A、X1A、F1A、G1A。
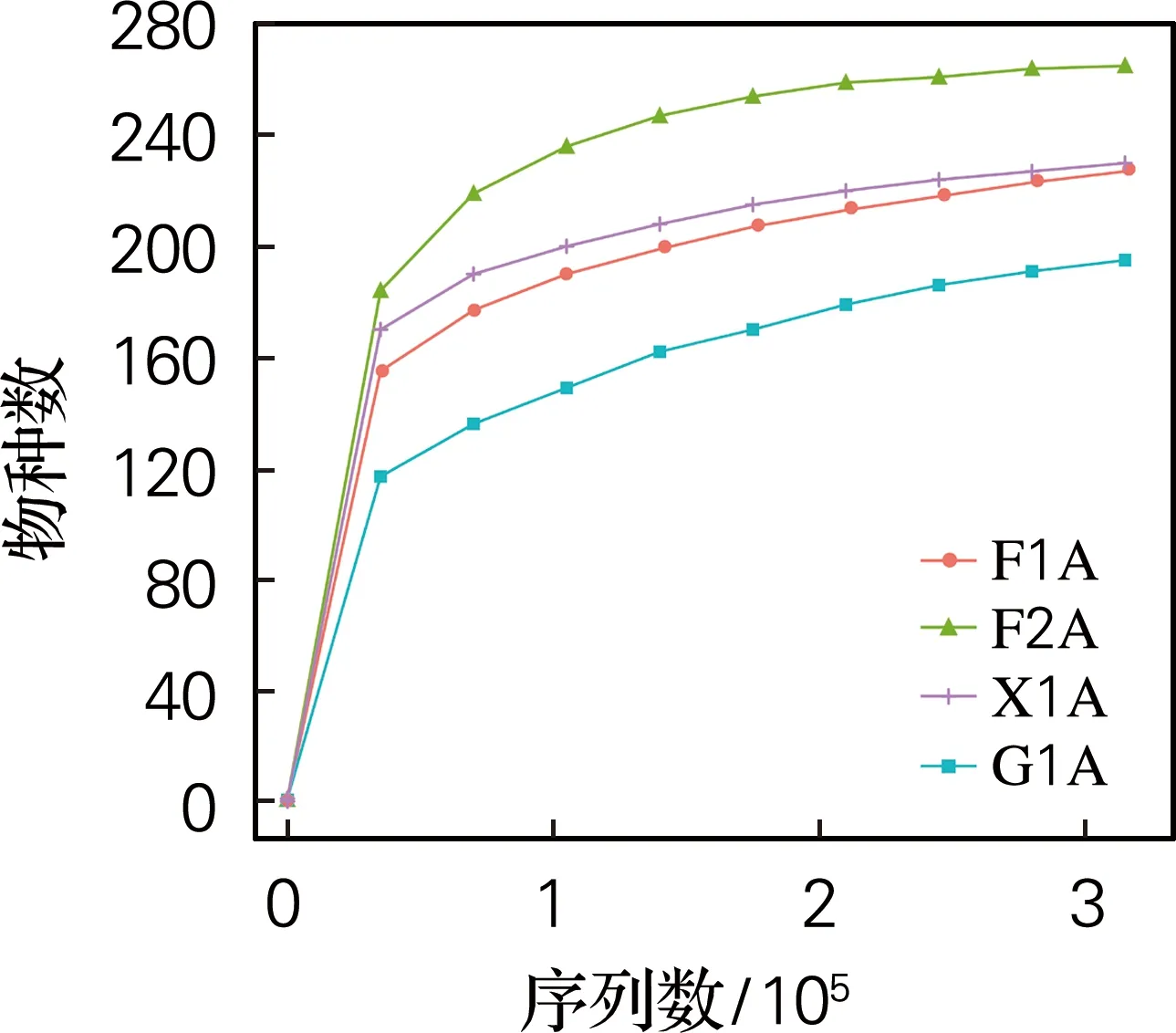
(a) 稀疏化曲线
在4个样本中,F2A样本的物种丰富度、物种均一性均最高,这可能是由于F2A样本所在培养基中CDM菌群已经生长了8 d,因此其中可能包含未降解纤维素以及纤维寡糖、纤维二糖、葡萄糖等纤维素降解中间产物,碳源的复杂程度高于纤维素降解3 d (F1A)、纤维二糖培养2 d (X1A)和葡萄糖培养2 d (G1A)的培养基,导致F2A样本的低丰度物种增加,菌种的多样性和均一性均增加,G1A样本的物种丰富度、物种均一性均最低,菌群CDM的多样性总体呈先高后低的趋势。有研究表明,物种的多样性与碳源复杂度成正相关[9]。对蜂粮样品进行菌群多样性分析时发现,蜂粮随着发酵时间的延长,其样本中菌群丰富度和多样性同样出现先升高在降低的趋势[10],与本研究结果一致。
从图2可以看出,4个样本共有148个OTU,约占OTU总数的47%,表明该CDM菌群稳定,这些OTU在整个纤维素降解过程中起着不可或缺的作用。各个样本中独有的OTU分别为F1A中1个、F2A中30个、X1A中6个、G1A中22个,说明CDM菌群在降解纤维素的过程中其菌群多样性不断变化,进一步证明了菌群组成是随着碳源复杂程度的变化而变化。
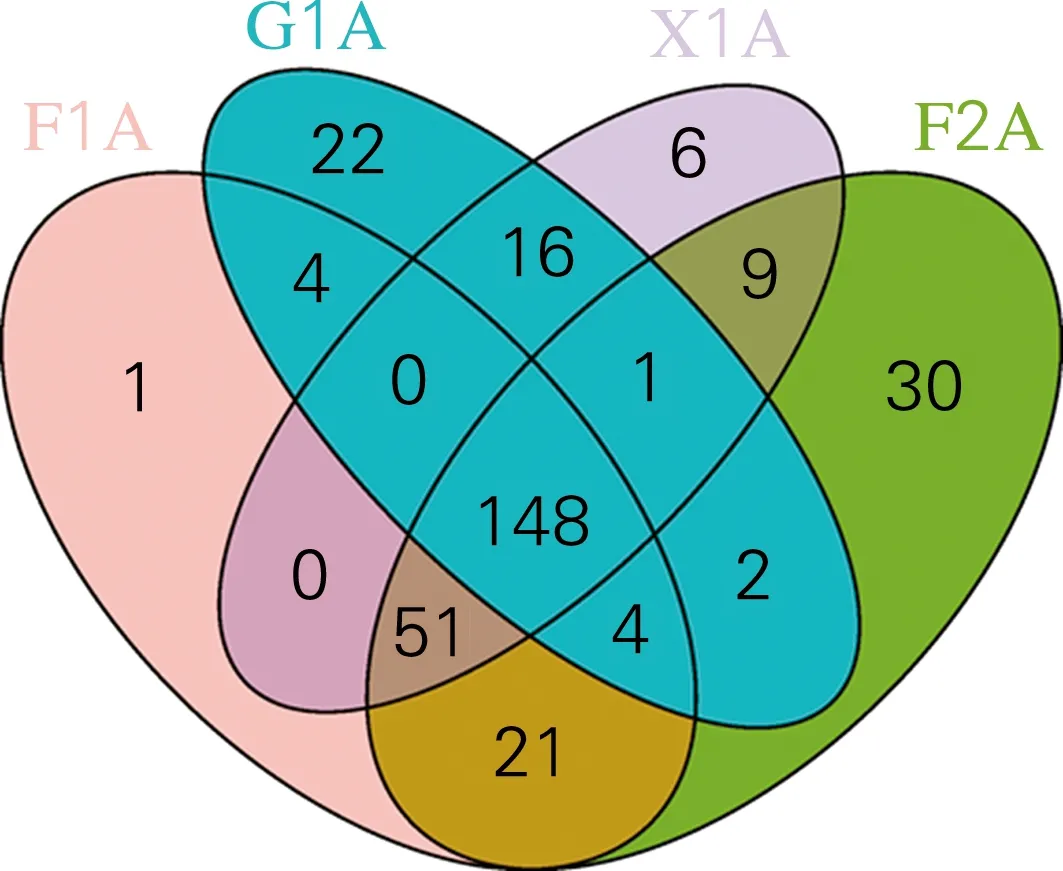
图2 OTU在各样本中分布的韦恩图
2.3 利用不同纤维素代谢产物为碳源时菌群CDM的β多样性比较
通过非加权UniFrac距离的β多样性来研究CDM在利用不同纤维素代谢产物为碳源时的菌群结构差异,结果如图3所示。从图3中可以看出,F1A与F2A之间距离较近,并且滤纸、纤维二糖和葡萄糖3种不同碳源之间样本彼此距离较远,因此推测在纤维素降解过程中,菌群结构差异随着碳源复杂程度的变化而变化。杨冰[11]分别以葡萄糖和黄水为碳源研究颗粒污泥中菌群结构变化时发现,其菌群结构受到碳源的影响有显著差异,与本实验结果研究一致。
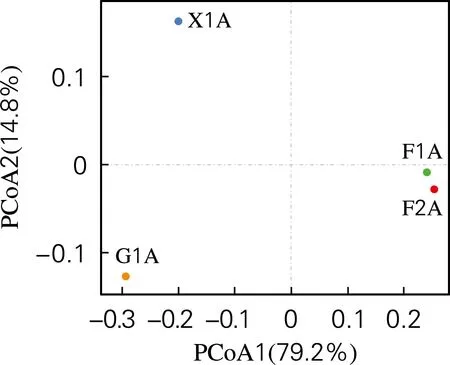
图3 菌群CDM基于非加权Unifrac距离的β多样性分析
2.4 利用不同纤维素代谢产物为碳源时菌群CDM在各个门水平上的动态变化
计算菌群CDM在细菌门水平的相对丰度,结果如图4所示。从图4可以看出,变形菌门(Proteobacteria)和厚壁菌门(Firmicutes)是所有样本中的主要门类,总相对丰度分别占68.53%和27.46%。有研究表明,在以竹子为食的大熊猫的肠道中发现变形菌门与厚壁菌门为优势门类[12-13],这与本研究结果相一致。F1A样本中主要门类为变形菌门和厚壁菌门,由此推测这两个门类共同参与纤维素降解的前期。在F2A、X1A样本中,变形菌门相对丰度依次降低,厚壁菌门相对丰度依次升高,由此可以发现变形菌门与厚壁菌门在纤维素降解的前中后期以此消彼长的动态变化协同降解纤维素。在G1A样本中主要门类为变形菌门,其相对丰度比F1A样本还高,由此可以看出变形菌门更倾向于在纤维素降解末期发挥作用。
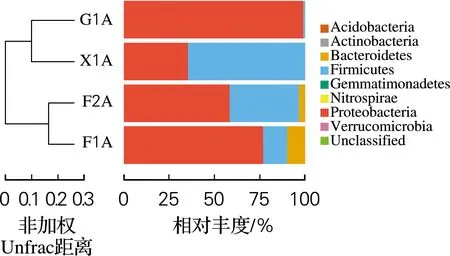
图4 细菌门水平相对丰度图
拟杆菌门(Bacteroidetes)仅在F1A、F2A样本中有发现,总相对丰度仅为3.97%,由此可以推测拟杆菌门也在前期和中期参与纤维素的降解。有研究证明拟杆菌门对碳水化合物具有较好的降解能力[14],在来自食草动物瘤胃、肠道以及堆肥等许多生物环境中均有发现[15-17]。另外,本研究还检出较低丰度的门。放线菌门(Actinobacteria)相对丰度为0.001%~0.012%,但在所有样本中均有发现,该门在一些纤维素降解菌群的相关研究中也常有发现[18]。
2.5 利用不同纤维素代谢产物为碳源时菌群CDM在各个科及属水平上的动态变化
对菌群CDM在利用不同纤维素代谢产物为碳源时科及属水平的群落结构进行分析。从图5中可以看出,所有样本中科主要包括草酸杆菌科(Oxalobacteraceae)54.22%、类芽孢杆菌科(Paenibacillaceae) 28.58%、产碱菌科(Alcaligenaceae) 12.97%,并含有少量丰度的噬纤维菌科(Cytophagaceae) 3.31%、芽孢杆菌科(Bacillacea) 0.37%和柄杆菌科(Caulobacteraceae) 0.15%。已有研究学者从瘤胃中分离出一种纤维素酶高产的产碱菌科纤维素分解菌[19-20]。4个样本中属主要包括草螺菌属(Herbaspirillum) 53.984%、少量的生孢噬纤维菌属(Sporocytophaga) 3.307%、科恩氏菌属(Cohnella) 3.147%,产碱杆菌属(Advenella) 2.221%、类芽孢杆菌属(Paenibacillus)1.941%、芽孢杆菌属(Bacillus) 0.373%、短波单胞菌属(Brevundimonas) 0.154%。
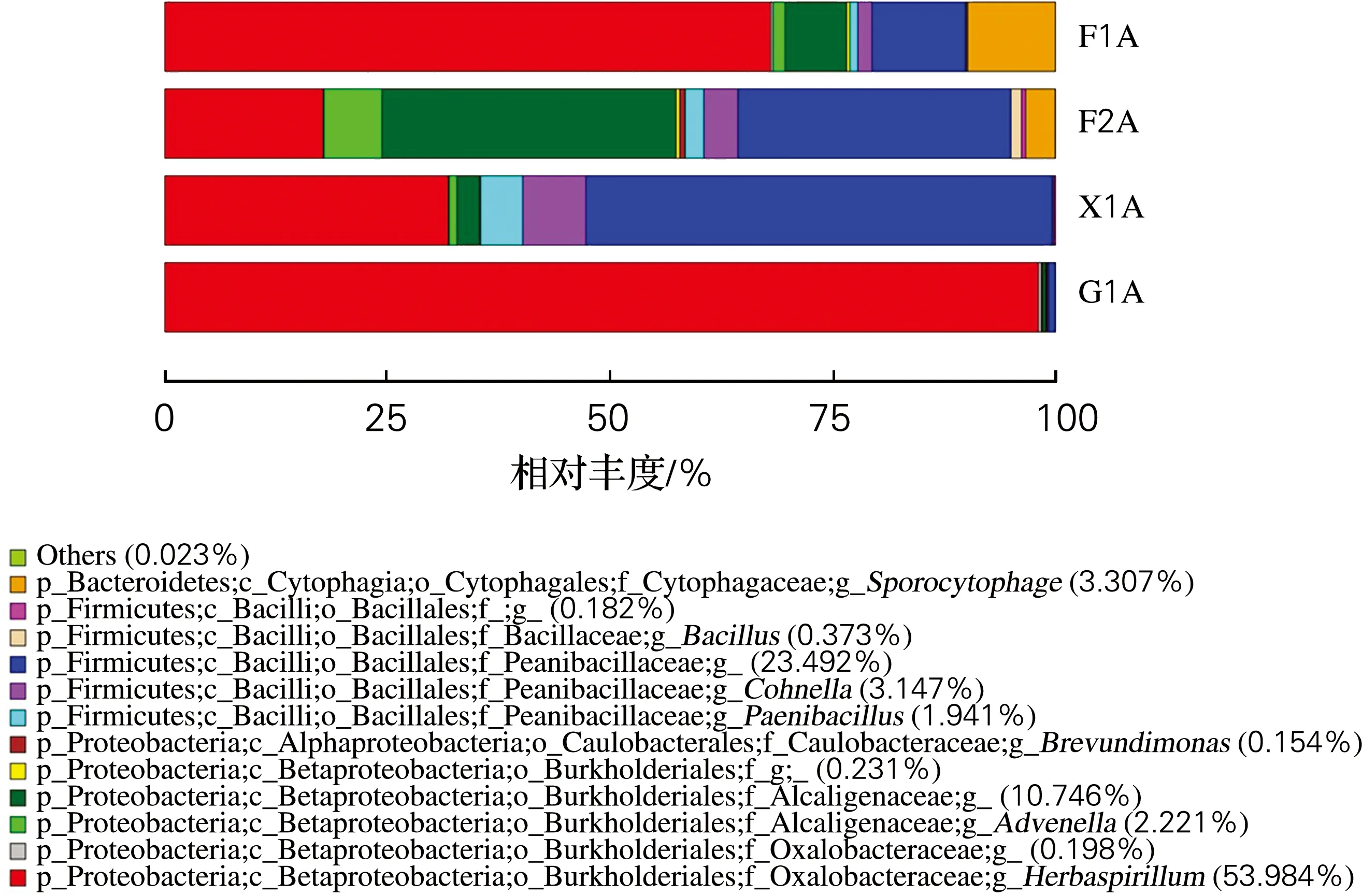
图5 菌群CDM在各个样本中细菌科及属的丰度分布统计图
根据菌群CDM在F1A、F2A、X1A和G1A 4个样本中的属水平相对丰度变化进而推测各属在纤维素降解过程中的主要作用时期,以及它们作用纤维素、纤维糊精/纤维寡糖、纤维二糖、葡萄糖4种底物的优先级顺序,如表2所示。
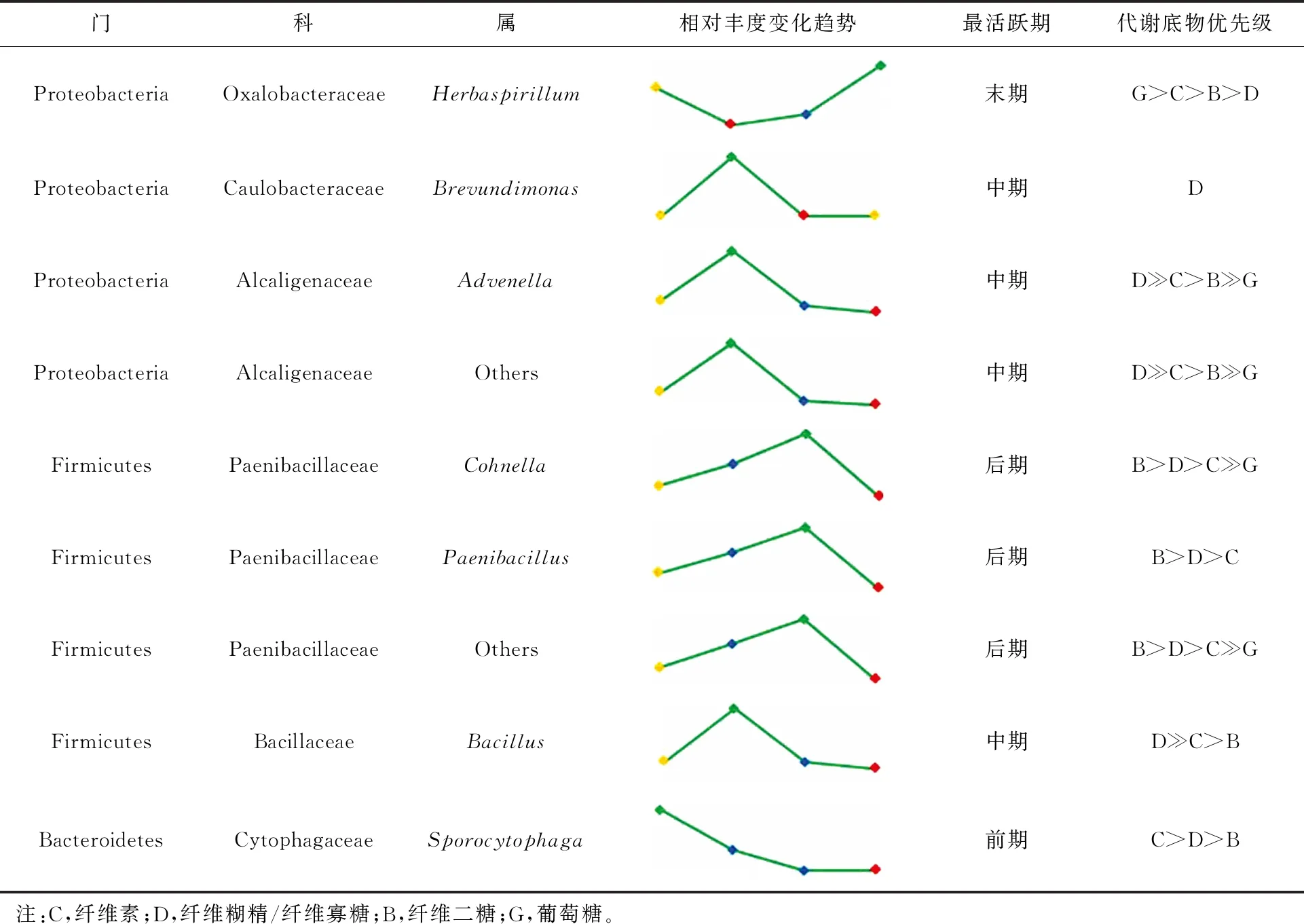
表2 菌群CDM中观察到的分类群及其功能预测
2.5.1 生孢噬纤维菌属(Sporocytophaga)
在F1A样本中相对丰度最高,F2A样本中相对丰度有所降低,X1A样本和G1A样本中未被观察到,有文献报道该属可以利用纤维素作为唯一的碳源和能源[21],故此推测生孢噬纤维菌属(Sporocytophaga)直接作用于纤维素。
2.5.2 短波单胞菌属(Brevundimonas)、芽孢杆菌属(Bacillus)和产碱杆菌属(Advenella)
在F2A样本中相对丰度最高,在X1A样本和G1A样本中相对丰度很低。Corno等[22]发现Brevundimonas可在纤维素培养基中的生长形成种群。Bacillus已被公认为纤维素酶的良好生产者,从堆肥、土壤和其他来源分离的许多Bacillus都能够有效分解纤维素和半纤维素[23]。Wang等[24]在对Proisotomaananevae肠道内纤维素降解菌筛选时获得产碱杆菌属(Advenella)。因此结合本研究结果推测短波单胞菌属(Brevundimonas)、芽孢杆菌属(Bacillus)和产碱杆菌属(Advenella)主要参与纤维素中期的降解。
2.5.3 科恩氏菌属(Cohnella)和类芽孢杆菌属(Paenibacillus)
相对丰度均在X1A样本中最高。有研究表明,Cohnella和Paenibacillus具备利用纤维素、半纤维素、纤维二糖以及葡萄糖的能力[25-26]。推测科恩氏菌属(Cohnella)与类芽孢杆菌属(Paenibacillus)倾向于降解后期对纤维二糖的降解。
2.5.4 草螺菌属(Herbaspirillum)
在4个样本中优势明显,姜乃文等[27]筛选出一株能够产纤维素酶的海洋杆菌,经菌种鉴定分析表明该菌株属于草螺菌属,并且对该属产生的纤维素酶的酶活力进行了测定。在G1A样本中草螺菌属(Herbaspirillum)的相对丰度达到最高(98.05%),且有研究证实该属可以用葡萄糖等碳源作为能源[28-29],因此推测该属倾向于在纤维素降解末期参与葡萄糖的代谢。
参与降解前中期的生孢噬纤维菌属(Sporocytophaga)、短波单胞菌属(Brevundimonas)、产碱杆菌属(Advenella)和芽孢杆菌属(Bacillus)与后末期的类芽孢杆菌属(Paenibacillus)、科恩氏菌属(Cohnella)和草螺菌属(Herbaspirillum)以此消彼长的动态变化协同降解。
3 结 论
本实验利用16S rRNA基因扩增子测序分析技术对一个纤维素降解菌群(CDM)在利用不同的纤维素代谢物为底物时物种组成及结构进行动态分析。α多样性分析表明菌群CDM的多样性与碳源复杂度成正相关,出现先升高后降低的变化趋势。β多样性分析表明菌群CDM的结构随着碳源复杂度的变化而变化。在门水平的相对丰度变化分析表明,变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)推测是参与纤维素降解过程的三大主要门类。在属水平,生孢噬纤维菌属(Sporocytophaga)、短波单胞菌属(Brevundimonas)、产碱杆菌属(Advenella)和芽孢杆菌属(Bacillus)推测主要参与纤维素降解前中期,类芽孢杆菌属(Paenibacillus)、科恩氏菌属(Cohnella)和草螺菌属(Herbaspirillum)推测主要参与纤维素降解后末期,两类菌属以此消彼长的动态变化协同降解纤维素。
猜你喜欢
河南医学研究(2022年19期)2022-10-19 00:44:18
环境工程技术学报(2022年3期)2022-06-05 07:20:20
昆钢科技(2021年6期)2021-03-09 06:10:20
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生态学报(2019年11期)2019-07-08 06:18:58
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
电源技术(2016年9期)2016-02-27 09:05:25
食品工业科技(2014年23期)2014-03-11 18:19:08
