
端丙炔酯基环氧乙烷四氢呋喃无规共聚醚的合成与性能研究
2022-09-02 02:05刘人天翟进贤杨荣杰宋振坤
火炸药学报 2022年4期
刘人天,翟进贤,杨荣杰,宋振坤
(北京理工大学 材料学院,北京 100081)
引 言
在固体复合推进剂配方中添加高能填料是提高其能量的重要途径;以端羟基预聚物为黏合剂、异氰酸酯化合物为固化剂进行交联反应是制备固体复合推进剂的典型方法。然而,由于异氰酸酯化合物与高能氧化剂二硝酰胺铵(ADN)的化学不相容性,制约了高能复合推进剂的发展。亚铜催化端炔基-叠氮基点击化学反应(CuAAC)具有反应条件温和、湿度不敏感、与ADN相容性好等优点,在非异氰酸酯固化交联制备高能复合推进剂方面获得广泛关注。[1-6]
利用叠氮聚醚主链上的叠氮甲基基团,Reshmi[7]以端羟基聚叠氮缩水甘油醚(GAP)为黏合剂、两官能度琥珀酸二丙炔酯(BPS)为固化剂,60℃下固化两天,通过叠氮与炔基环化反应得到一系列聚三唑交联GAP热固性弹性体。Li[8]以端羟基聚3-甲基-3-叠氮甲基氧丁环(PAMMO)为黏合剂、1,4-环己烷二甲酸丙炔酯为固化剂,50℃下放置4天,制得聚三唑交联PAMMO热固性弹性体。由于叠氮聚醚与双官能度炔基固化剂属无规交联反应,所得上述弹性体力学性能不佳。Menke、Rahm[9-10]在GAP/BPS体系中加入三羟甲基乙烷三硝酸酯增塑剂,同时添加HMX、ADN等含能固体填料,通过端炔基/叠氮基环化反应得到聚三唑聚叠氮聚醚固体复合推进剂,但力学性能仍不理想。为改善GAP/BPS体系力学性能,Kristensen[11]借助GAP预聚物中同时含羟基、叠氮基的特点,采用双炔基固化剂和异氰酸酯固化剂,使炔基与叠氮基反应、异氰酸酯与端羟基进行反应,得到含奥克托今固体填料的无烟固体复合推进剂,该体系延伸率有所提高,但玻璃化温度较高。
叠氮聚醚与炔基固化剂无规交联难以满足固体复合推进剂力学性能要求,通过聚三唑末端交联、制备网络结构规整的弹性体是改善其力学性能的主要途径。曲正阳等[12-13]分别以端羟基聚乙二醇400、端羟基聚环氧乙烷-四氢呋喃共聚醚(PET)为预聚物前体,四氢呋喃为溶剂,同时添加丙炔溴、叔丁醇钾,制备得到了端炔基聚乙二醇(PTPEG)和端炔基聚环氧乙烷-四氢呋喃共聚醚(PTPET);分别将PTPEG、PTPET与叠氮固化剂混合,亚铜催化剂存在下制备得到末端交联、力学性能良好的聚三唑聚醚弹性体。翟进贤等[14]则将PET预聚物进行端基叠氮化反应,制备得到端叠氮基聚环氧乙烷-四氢呋喃共聚醚(ATPET),随后将ATPET与三炔丙基胺混合,在亚铜催化剂作用下同样制备得到末端交联、力学性能良好的聚三唑交联聚醚弹性体。由于亚铜化合物易发生氧化或歧化反应而失去催化效果,上述末端交联反应在实际应用中具有一定的局限性。
提高炔基或叠氮基反应活性,是降低亚铜催化剂依赖、实现炔基与叠氮基非催化反应制备聚三唑末端交联弹性体的又一前进方向。Pascoal等[15]研究表明,若在炔基官能团附近引入拉电子基团可提高其与叠氮基的反应速率。Patrick等[16]通过竞争反应研究了拉电子效应对炔基化合物反应活性的影响,并且借助密度泛函理论研究了环境化学对炔基/叠氮基环加成反应影响的定量规律。利用羰基拉电子效应,Earla[17]的研究表明炔基单酯、双酯化合物在室温下即可与叠氮基团反应生成三唑化合物。Higa[18]的研究表明,磺氧基取代的炔烃化合物与叠氮基在室温下也可快速反应生成三唑化合物。Qin等[19]将双(芳酰基乙炔)和二叠氮化物在极性溶剂中混合,非催化条件下制备得到重均相对分子质量26700的聚芳酰基三唑化合物。可以看出,对炔基基团进行化学环境修饰是提高其与叠氮基化学反应效率的一条重要出路,而将该方法应用于聚合物末端改性、以及非催化条件下制备聚三唑末端交联弹性体的研究还未见报道。
为克服因亚铜催化剂不稳定、易失活而影响叠氮基/炔基反应彻底性的不足,本研究以数均分子质量4000g/mol的端羟基聚环氧乙烷-四氢呋喃共聚醚为反应底物,通过端基酯化反应制备得到了炔基反应活性较高的端丙炔酯基聚环氧乙烷-四氢呋喃共聚醚预聚物(propiolate-terminated ethylene oxide-tetrahydrofuran copolyether,PTPET),并对其性能进行了表征,以期为非催化条件下制备聚三唑交联聚醚固体复合推进剂原料提供参考。
1 实 验
1.1 试剂与仪器
端羟基环氧乙烷-四氢呋喃无规共聚醚 (PET,Mn=4038g/mol,羟值为0.435mmol/g)、叠氮缩水甘油醚齐聚物GAP (Mn=480g/mol,平均官能度为3.82),洛阳黎明化工研究院;丙炔酸,纯度90%,上海迈瑞尔化学技术有限公司;二环己基碳二亚胺(DCC),纯度99%,阿拉丁试剂(上海)有限公司。
Nicolet 6700红外测试仪,美国热电公司;AVANCE DRX-500核磁共振波谱仪,德国布鲁克公司;Waters Breeze TM2 HPLC System GPC凝胶渗透色谱测试仪,美国Waters公司;Rheostress 300扭矩流变仪,德国Thermo Haake公司;CMT4104电子拉力试验机,MTS公司。
1.2 实验方法
室温下,向装有机械搅拌装置的250mL三口烧瓶中依次加入20g PET、3.09g二环己基碳二亚胺、100mL二氯甲烷,机械搅拌至完全溶解。然后逐滴加入1.17g丙炔酸,室温下反应8h,混合物由淡黄色变为红棕色。用饱和NaCl水溶液对反应混合物萃取2次,保留下层红棕色澄清液,加入无水硫酸钠除水,将液相分离后减压蒸馏除去二氯甲烷溶剂,真空干燥,得到红棕色黏稠状液体端丙炔酯基聚环氧乙烷-四氢呋喃共聚醚PTPET 16.8g。依据PET添加量,所得PTPET产率为84%。
1.3 弹性体的制备
分别制备固化参数为0.8~1.2(叠氮基与炔基的摩尔比)的PTPET/GAP弹性体。将PTPET黏合剂与GAP叠氮固化剂按不同官能团摩尔比混合均匀,倒置于聚四氟乙烯模具中,真空除去气泡。然后,置于65℃恒温箱中固化,至混合基体中的叠氮基/炔基红外吸收峰完全消失,得到反应完全的聚三唑交联聚醚弹性体样品。
1.4 性能测试与表征
使用FTIR红外测试仪对样品进行红外扫描,扫描范围500~4000cm-1,扫描次数32次;利用核磁共振波谱仪对样品进行核磁13C谱分析,以TMS为内标,CDCl3为溶剂,使用反转门控去耦对PET和PTPET进行定量测试;将样品配成10~15mg/mL的四氢呋喃溶液,静置过夜,以四氢呋喃为流动相,在凝胶渗透色谱测试仪上进行聚合物相对分子质量测试;利用Rheostress 300扭矩流变仪分析聚合物黏度,剪切速率为1~10s-1,每秒采集一次,采集60s;按照GB9865将样品制成哑铃状,通过电子拉力试验机在室温下对其拉伸力学性能进行测试,拉伸速率10mm/min。
2 结果与讨论
2.1 酯化原理
利用DCC进行酯化反应的机理如图1所示。反应初始阶段,丙炔酸中的活泼氢向DCC中的氮原子进攻,形成中间体。之后,端羟基聚醚上的醇羟基氧原子进攻中间体上的羰基,形成丙炔酸酯化合物。DCC则生成不溶于二氯甲烷溶剂的二环己基脲沉淀,促使反应彻底进行,得到红棕色目标产物端丙炔酸酯基环氧乙烷-四氢呋喃无规共聚醚PTPET。
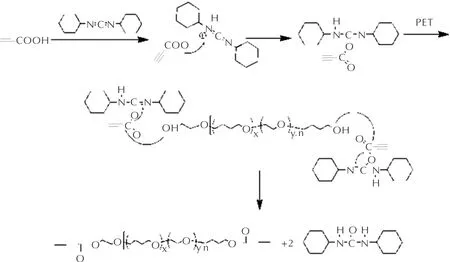
图1 PTPET酯化反应机理Fig.1 Esterification mechanism of PTPET
2.2 结构表征
2.2.1 FT-IR分析
对原料PET和产物PTPET进行红外光谱测试,其红外光谱图如图2所示。

图2 PET与PTPET的红外光谱图Fig.2 FTIR spectra for PET and PTPET
由图2可以看出,1716cm-1处为丙炔酸酯中C═O伸缩振动吸收峰,2116cm-1处为炔基基团C≡C伸缩振动吸收峰,3211cm-1处为端炔基结构中≡C—H的伸缩振动吸收峰。与PET红外吸收峰相比,PTPET在3470cm-1处的吸收峰完全消失,表明PET结构中的端羟基已完全酯化,生成了端丙炔酸酯基基团。
2.2.213C NMR 分析
为进一步确认PTPET酯化产物分子结构,对PET和PTPET进行13C核磁测试,所得13C-NMR谱图如图3所示。由图3可知,PET的13C-NMR谱图中,化学位移70.04~71.12处对应于测点 1和测点 2碳原子, 26.23对应于测点 3碳原子,72.55对应于测点 4碳原子。值得注意的是,与端羟基相连碳原子(测点 5和测点 6)对应于61.72和61.65处共振峰,至77.0处三重共振峰源于溶剂CDCl3。与PET谱图相比,在PTPET谱图中,测点 1、测点 2和测点 3碳原子共振峰位置保持不变,仍处于原化学位移处。由于端羟基酯化,测点 4、测点 5和测点 6碳原子共振峰分别移至71.1、74.53处。同时在152.3、152.1处出现测点 7和测点 8碳原子共振峰,76.0、75.1处出现测点 9和测点 10碳原子共振峰。由局部放大图可以看出,PET中的端羟基在PTPET谱图中完全消失,表明该酯化反应完全,生成了端丙炔酸酯基环氧乙烷-四氢呋喃无规共聚醚PTPET。

图3 PET与PTPET核磁13C谱图Fig.3 13C-NMR spectra for PET and PTPET
2.2.3 GPC分析
为确认PET酯化反应所得PTPET预聚物相对分子质量,对PET和PTPET预聚物进行凝胶渗透色谱测试(GPC),表1是其GPC参数测试结果。

表1 PET和PTPET的GPC结果参数Table 1 GPC parameters for PET and PTPET
由表1可以看出,PET预聚物的数均分子质量为4038g/mol,重均分子质量为6978g/mol,多分散指数为1.73;PTPET预聚物的数均分子质量为4153g/mol,重均分子质量为7177g/mol,多分散指数为1.73,PET与PTPET两者相对分子质量参数非常接近。
图4为PET与PTPET预聚物GPC流出曲线。由图4可以看出,PET与PTPET的GPC流出曲线位置和峰型基本一致。结合表1测试结果可以推断,PET端羟基与丙炔酸酯化过程中,仅仅是PET预聚物的端羟基发生了酯化,生成了PTPET,而聚合物并未发生断链、扩链等副反应。PTPET与PET的端基官能团含量相一致。

图4 PET与PTPET的GPC曲线Fig.4 GPC curves for PET and PTPET
2.3 黏度分析
PTPET黏度是其重要参数之一,直接影响其使用性能。为揭示端羟基丙炔酸酯化对共聚醚黏度的影响,利用Haake流变仪对PET和PTPET预聚物黏度进行了测试,图5为其剪切速率—黏度关系曲线。由图5可以看出,相同剪切速率下,PTPET预聚物黏度显著高于PET。鉴于预聚物PTPET与PET两者的相对分子质量及其分布极为接近,两者的唯一区别源于其端基官能团,因此PTPET预聚物的高黏度特性应源于其分子链的端位基团。对于丙炔酸酯基基团,羰基基团与炔基的三键可形成p轨道电子共轭结构,由于羰基结构中氧原子的拉电子效应,使得端炔基结构中的氢原子更加活泼,可与其他丙炔酸酯基上的羰基基团形成氢键[20],增加了PTPET分子链间的相互作用,PTPET分子链段运动阻力增加。因此,PTPET聚合物黏度显著高于PET体系。

图5 PET与PTPET剪切速率—黏度关系曲线Fig.5 Shear rate—viscosity curves for PET and PTPET
2.4 端炔基含量的确定
PTPET端炔基含量是确定固化剂GAP添加量、调节固化参数、获得不同网络交联结构弹性体的关键参数。目前,有关聚合物端炔基含量的定量测定方法尚未见文献报道,而通过聚合物前驱体官能团等价换算是确定聚合物端炔基含量的一种简洁、快速的方法。鉴于PTPET预聚物分子链结构中端羟基被完全酯化(见13C-NMR分析),且PET聚合物分子链在酯化过程中未发生断链、交联等副反应(见GPC分析),因此,可以推断PTPET聚合物中的端炔基摩尔数与PET中端羟基摩尔数相同,依据式(1)可计算获得PTPET预聚物的端炔基摩尔含量。
CAlkyne=CHydroxyl/(1+CHydroxyl×52)
(1)
式中:CAlkyne为PTPET中炔基的含量,mmol/g;CHydroxyl为PET中羟基的含量,mmol/g;52为丙炔酯基摩尔质量69g/mol与羟基摩尔质量17g/mol的差值,g/mol。
将产物PTPET前驱体PET羟值(0.435mmol/g)代入式(1),计算得PTPET的端炔基值为0.425mmol/g。
2.5 弹性体力学性能分析
依据上述2.4节确定的PTPET炔基值,将其与GAP固化剂按不同比例混合,制备不同R值(GAP中叠氮基与PTPET中炔基比例)的聚三唑交联聚醚弹性体。表2是该弹性体室温下力学性能测试结果。

表2 聚三唑交联弹性体的室温力学性能Table 2 Mechanical properties of polytriazole elastomers at room temperature
由表2可以看出,在R值为1时,弹性体拉伸强度为0.89MPa,断裂伸长率为189%,在5组弹性体中具有良好的力学性能。
3 结 论
(1)以PET为原料,通过端羟基酯化反应制备得到了端丙炔酯基环氧乙烷-四氢呋喃无规共聚醚目标化合物PTPET。FTIR、13C-NMR分析表明,PTPET分子结构中与端羟基有关的红外特征吸收峰和核磁共振峰完全消失,产物丙炔酸酯化反应完全。
(2)PTPET具有与前驱体PET相近的相对分子质量及其分布特性。相近相对分子质量条件下,PTPET较PET具有较高的黏度。
(3)非催化条件下,PTPET与GAP可交联固化形成聚三唑交联弹性体,力学性能良好,为制备聚三唑聚醚复合推进剂拓宽了原料基础。
猜你喜欢
现代园艺(2022年17期)2022-08-23
汽车实用技术(2021年10期)2021-06-04
中国科技纵横(2019年23期)2019-02-14
首都体育学院学报(2018年5期)2018-12-04
计算机辅助工程(2018年2期)2018-06-03
中学课程辅导·教学研究(2017年29期)2018-02-26
城市建设理论研究(2012年6期)2012-04-10
城市建设理论研究(2011年23期)2011-12-20
数理化学习·高一二版(2009年1期)2009-03-19
中国科技术语(2004年4期)2004-03-18
