
金属氧化物在OX-ZEO催化剂中催化COx加氢制低碳烯烃研究进展
2022-08-29 04:09张鹏孟凡会杨贵楠李忠
化工进展 2022年8期
张鹏,孟凡会,杨贵楠,李忠
(太原理工大学省部共建煤基能源清洁高效利用国家重点实验室,山西太原 030024)
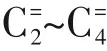
采用金属氧化物-分子筛(OX-ZEO)双功能催化剂催化CO(CO/CO)加氢转化直接制低碳烯烃技术是近几年的研究热点。该技术将CO加氢制甲醇和甲醇制烯烃两步反应耦联起来,使C—O活化和C—C 耦合分别控制在不同的活性位点,实现了一步法由合成气到低碳烯烃的高选择性转化,其示意图见图1。在OX-ZEO 双功能催化剂中,CO首先在金属氧化物上活化并形成乙烯酮、甲氧基、甲醇等活泼含氧中间体,含氧中间体迅速扩散进入酸性分子筛(SAPO-34、MOR、SAPO-18、SSZ-13、RUB-13等)生成低碳烯烃。由于CO/H的吸附和活化均发生在金属氧化物上,因而金属氧化物主要决定反应的催化活性,分子筛则因其特定的孔道结构和酸性质决定目标产物的类型和分布。近年来,分子筛在双功能催化剂中催化CO/CO加氢取得了重要进展。本文针对OX-ZEO中的金属氧化物,概述了金属氧化物在OX-ZEO中催化CO加氢制低碳烯烃反应中的研究进展,重点讨论了包括金属氧化物种类和组成、金属氧化物制备方法、金属氧化物与分子筛“亲密度”对反应性能的影响,探讨了催化反应机理、氧空位的作用及抑制副反应的策略,分析了OX-ZEO催化反应面临的问题和挑战,并对OX-ZEO催化剂中金属氧化物的设计进行了展望。
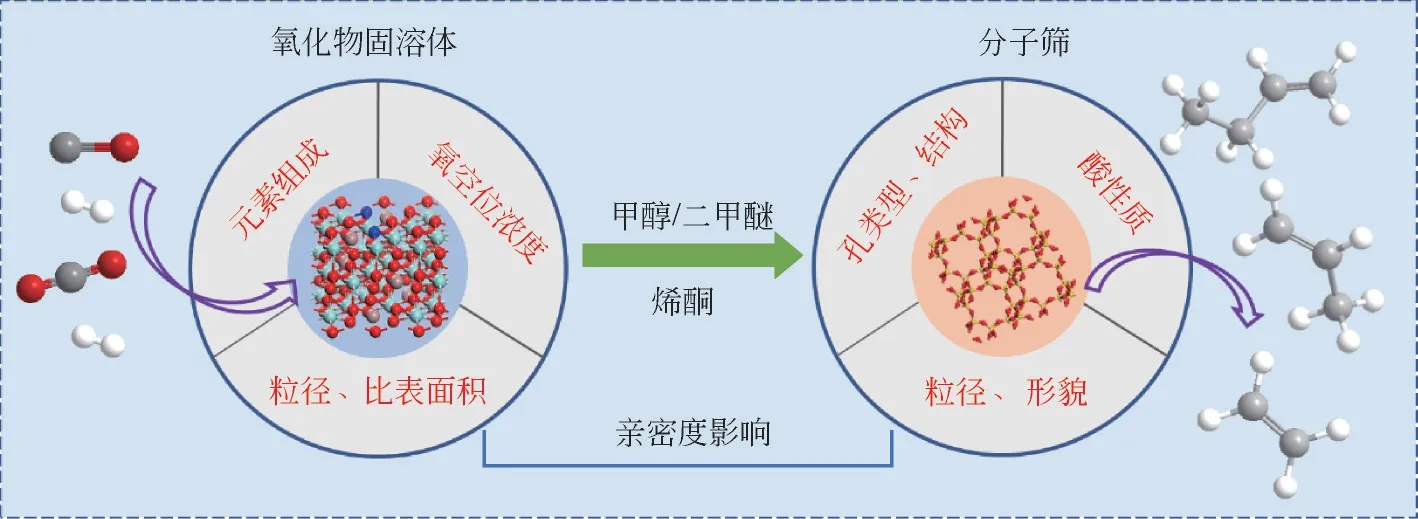
图1 COx加氢经OX-ZEO双功能催化剂合成低碳烯烃示意图以及影响性能关键因素
1 热力学分析
OX-ZEO过程可以看作甲醇合成反应和甲醇制烯烃(MTO)反应的耦合,由于MTO 反应在低温下(<350℃)催化活性低,因此OX-ZEO催化剂反应温度一般在350℃以上,然而合成甲醇反应是放热反应(CO+2H=== ===== CHOH,Δ=-90.4kJ/mol;CO+3H=== ===== CHOH+HO, Δ= - 49.4kJ/mol),热力学因素决定了低温、高压有利于甲醇的生成,采用HSC 6.0软件对合成气制甲醇/乙烯以及CO加氢制甲醇/乙烯的热力学进行了分析,见图2。从图2(a)可以看出,合成气制甲醇反应在压力3.0MPa、温度350~400℃范围内时,CO转化率仅为2.0%~0.5%,通过耦合分子筛可以实现甲醇等含氧中间产物的快速转化,拉动反应不断正向进行,进而提高催化活性。由图2(b)可知,CO 直接转化制低碳烯烃在温度350~400℃、压力3.0MPa时的平衡转化率高达93.9%~80.6%。
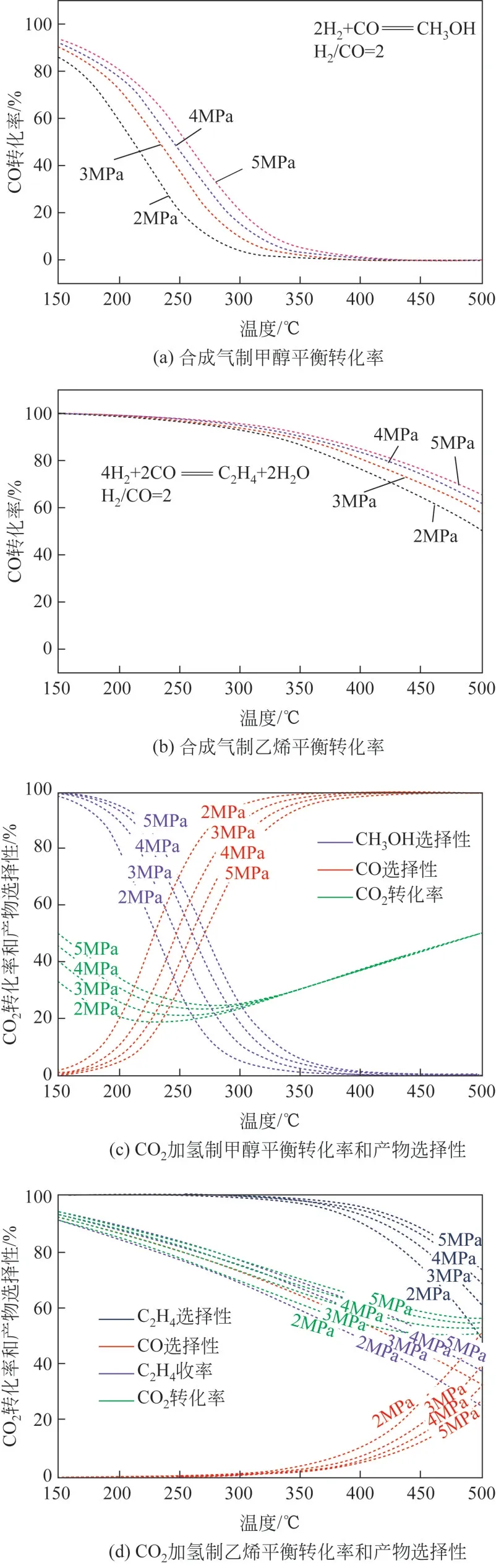
图2 合成气制甲醇/乙烯以及CO2加氢制甲醇/乙烯平衡转化率和组成
2 金属氧化物类型
由于甲醇合成反应与MTO 反应的最佳反应温度并不匹配,必须对双功能催化剂进行设计和调控,以使这两个反应具备更好的“热耦合”性。Fujimoto 等最早尝试将甲醇催化剂PdSiO与Y 型分子筛结合用于合成气转化,但是产物主要以低碳烷烃为主,典型的Cu-Zn-Al 甲醇催化剂在高温下活性及稳定性差、甲醇选择性低。近年来,尖晶石或固溶体结构的氧化物如ZnCrO、ZnZrO、InZrO、GaZrO等逐渐受到研究者的关注,此类催化剂不仅表现出极好的高温甲醇合成活性和稳定性,其耦合SAPO-34后同样表现出较优的CO加氢转化制烯烃性能,具有潜在的工业应用前景。一般来说,适用于OX-ZEO催化剂的氧化物组分需具备双活性中心,一种活性中心活化CO,另一种活性中心可以解离活化H,促进CO加氢生成含氧中间体。MO(M=Zn、In、Ga)为d电子构型氧化物,M中d电子通过与H配位可以实现对H的异裂解离(H-H),因此Zn、In、Ga元素通常作为活化H的活性中心。目前,氧化物研究的热点主要针对Zn基、In基和Ga基氧化物改性,从而提高催化性能。
2.1 Zn基氧化物
ZnO 解离H能力较强,但其活化CO能力较差,通过掺杂Cr、Zr、Al、Mn等元素可以调控表面氧空位含量和H解离能力,协同活化CO和H,促进CO加氢反应活性。2016 年,Jiao 等首次报道了ZnCrO氧化物耦合SAPO-34 分子筛用于合成气直接转化制低碳烯烃(STO)反应,并命名为OX-ZEO 过程,如图3 所示。在反应温度400℃、H/CO=1.5、2.5MPa 反应条件下,CO 转化率达17%,低碳烯烃选择性高达80%,选择性远高于费托反应过程所报道的最大值61%,且催化剂保持了良好的稳定性。Cheng 等将ZnZrO氧化物与SAPO-34分子筛耦合,在反应温度400℃、反应压力1.0MPa、H/CO=2 条件下,获得了11%的CO转化率,低碳烯烃选择性达到70%。Liu等发现,具有尖晶石结构的ZnAlO氧化物复合SAPO-34后,在CO转化率达到24%时,低碳烯烃选择性达到80%。ZnZrO氧化物在OX-ZEO催化体系中研究较为广泛,但其活性普遍较低,通过进一步引入Ce、In、La等助剂元素修饰可进一步增加表面氧空位含量,提高催化性能。本文作者课题组Meng等发现,采用Ce掺杂Zr-Zn制备的三元氧化物与SAPO-34 结合,在催化STO 反应中可使CO转化率达到25.6%,低碳烯烃选择性达到78.6%。Wang 等发现,在ZnZrO固溶体中掺杂Ce 制备的ZnCeZrO与SAPO-34 复合,在温和的反应条件300℃、0.1MPa 下,可使低碳烯烃选择性高达83%,副产物CO选择性降为6%,但CO 转化率仅为7%。通过DFT 理论计算表明,Ce 插入到ZnZrO中提高了HO 的解离自由能以及羧酸基(HOCO)的生成自由能,抑制了水汽变换(WGS)反应的进行,因而降低了CO选择性。
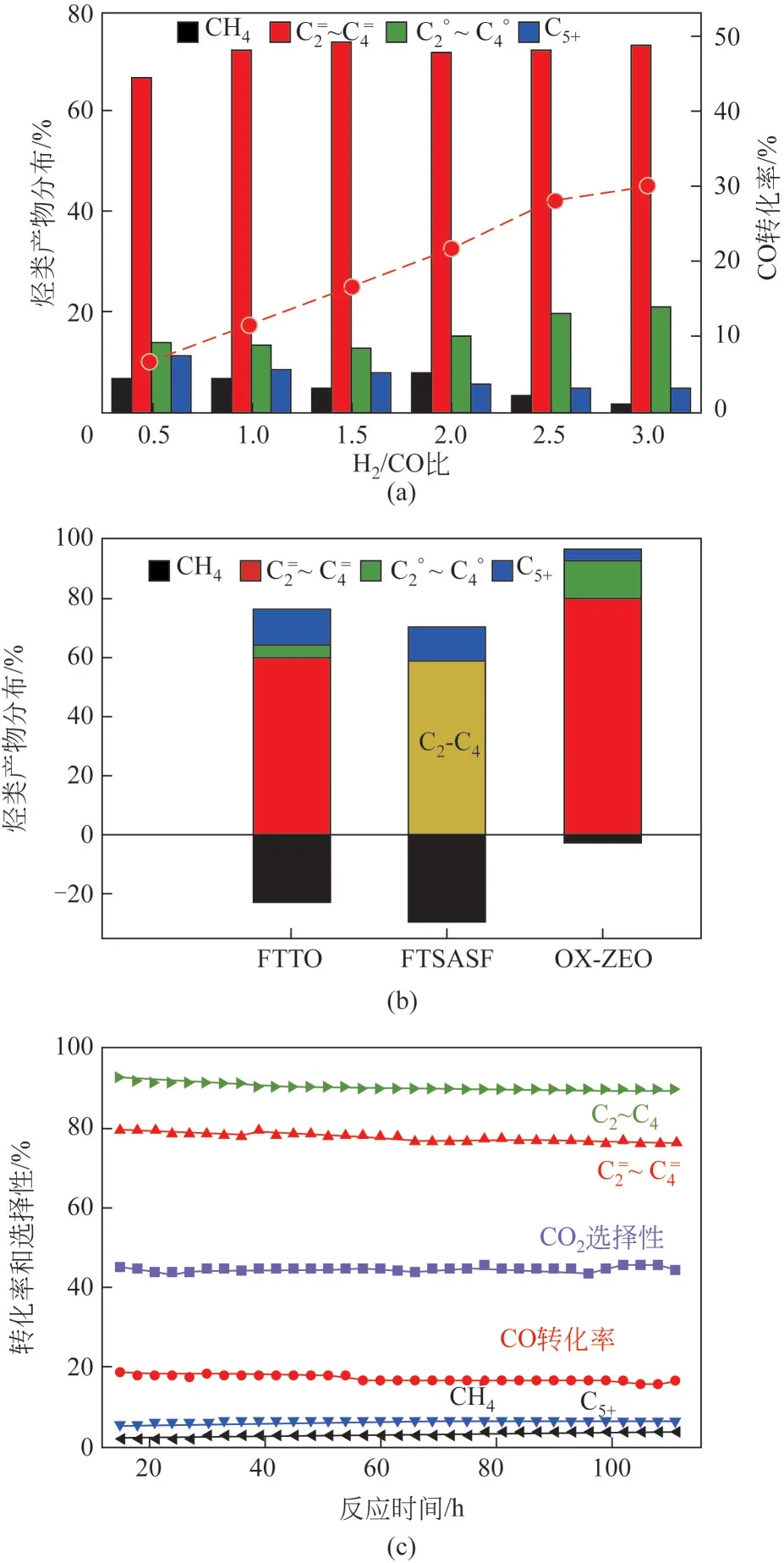
图3 ZnCrOx/SAPO-34催化剂在不同H2/CO比条件下的催化性能及稳定性评价[13]
得益于OX-ZEO路线在STO反应中取得的巨大突破,研究人员将OX-ZEO路线扩展应用到CO加氢方向。Li等将ZnZrO与SAPO-34耦合的双功能催化剂用于CO加氢反应,如图4 所示,在CO转化率为12.6%时,碳氢化合物中低碳烯烃的选择性为80%,副产物CO 选择性为47%,机理研究表明CHO(CHO、CHO、CHOH)为连接ZnZrO与SAPO-34的反应中间产物。Liu等采用ZnAlO氧化物复合SAPO-34 后,在CO转化率达到15%时,低碳烯烃选择性达到87%。Mou 等发展了ZnOMnO氧化物复合SAPO-34 的双功能催化剂催化CO加氢制低碳烯烃,在CO转化率为30%时,低碳烯烃选择性为80.2%,低碳烯烃收率高达10.7%。Zhang 等设计了La 修饰的ZnZrO,发现La 的引入可产生更多的氧空位,促进CO活化以及甲酸根和甲氧基中间体的生成,在相同反应条件下CO加氢性能优于未经La 修饰的ZnZrO。此外,也有文献报道采用ZnO-YO复合SAPO-34 的双功能催化剂也可用于CO加氢制低碳烯烃反应,在CO转化率达到27.6%时,烃类中低碳烯烃选择性达到83.9%,但是副产物CO 选择性较高,为85.0%。
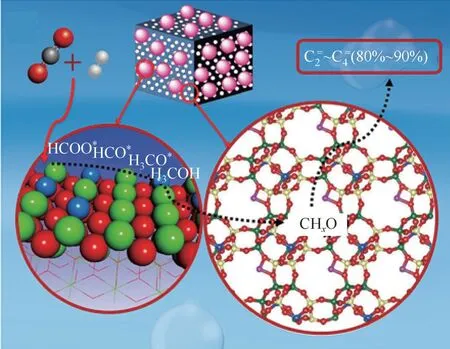
图4 ZnZrOx/SAPO-34双功能催化剂催化CO2加氢制低碳烯烃示意图[30]
2.2 In基氧化物
InO易产生氧空位,具有良好的CO和H活化能力,被认为是CO加氢高温制甲醇最理想的催化剂之一,通过引入Zr 可进一步增加甲醇收率。但单一InO并不具备CO 加氢活性,主要原因是InO在CO 气氛中350℃下就容易被完全还原成金属In,因此In 基双功能催化剂多用于CO加氢转化制低碳烯烃方向的研究。Gao 等通过沉积沉淀法制备了InO/ZrO复合氧化物,并与SAPO-34 分子筛耦合用于CO加氢制低碳烯烃反应,在CO转化率达到20%时,烃类产物中乙烯和丙烯总选择性达到80%~90%,然而副产物CO 选择性高达85%。Dang 等和Gao 等研究了在InO中掺杂Zr的作用,证实Zr的掺杂提高了In-Zr复合氧化物的氧空位浓度和抗烧结能力,有助于稳定中间产物,通过调控Zr/In 原子比,在保持低碳烯烃选择性为65%~80%的同时,CO转化率可达15%~27%,然而副产物CO选择性高于60%,进一步采用Zn助剂修饰则可将CO选择性降至47%。
对比单一InO,掺杂Ce、Cr 元素有助于产生更多的表面氧空位,可提高CO加氢催化活性和甲醇选择性。Wang 等通过DFT 理论计算发现,Ce 和Cr插入到InO晶格后提高了HCOO中间体与InCeO和InCrO表面的电子相互作用,降低了生成HCOO和CHOH 中间体的自由能,因而提高了中间产物甲醇的生成速率,且对比InCeO,InCrO表现出更优的CO加氢性能;将InCrO与SAPO-34结合,在反应温度350℃、反应压力3.5MPa时,可使CO转化率达到33.6%,低碳烯烃选择性为75.0%,副产物CO选择性为55.0%。
2.3 Ga基氧化物
GaO可以在高温下解离H,且在一定温度范围内,温度越高,H的解离能力越强,在甲醇合成反应中,Ga 主要作为助剂来提高活性组分如Cu物种或Zn物种的分散,改善加氢能力,其作为活性氧化物组分用于OX-ZEO催化剂的研究目前报道较少。本文作者课题组Zhang 等采用氧空位含量丰富的MnO活化CO,通过引入Ga 物种实现了对H解离能力的精准调控,既有助于提高催化活性又有效避免了烯烃的二次加氢,设计的GaMnO/SAPO-34 双功能催化剂在CO 转化率为13.7%时,低碳烯烃选择性高达88.3%。为进一步提高催化活性,通过掺杂Cr调控CO活化能力,发现Cr的引入提升了高温下CO 活化并转化为HCOO物种的能力,使C—O活化与C—C耦合的温度更加匹配,设计的CrMnGaO/SAPO-34 低碳烯烃选择性达到87%时,CO转化率高达43.5%。
GaO复合ZnO形成的ZnGaO尖晶石结构具有较高的氧空位含量。Liu 等将具有尖晶石结构ZnGaO耦合SAPO-34 用于催化CO加氢转化制低碳烯烃,在CO转化率达到30%时低碳烯烃选择性为77%,在CO转化率达到13%时低碳烯烃选择性为86%。最近的研究发现,GaZrO固溶体结构在催化CO加氢制甲醇反应中表现出极好的高温活性和选择性。本文作者课题组Zhang 等设计了GaZrO氧化物耦合SAPO-34 双功能催化剂,获得了26.7%的CO转化率,低碳烯烃选择性为88.8%,副产物CO 选择性仅为52.4%,红外表征结合DFT理论计算表明,Ga 与Zr 之间的协同作用有助于在高温下生成和稳定CHO中间体,抑制RWGS 反应,从而降低副产物CO的量。
2.4 其他氧化物
除以上所报道的尖晶石或固溶体结构的氧化物,单一活性氧化物如MnO、ZrO、CrO以及复合氧化物如CeZrO、Mo-ZrO等也可应用于OX-ZEO 双功能催化体系,在CO加氢制低碳烯烃或芳烃等研究中受到广泛关注。
综上所述,Zn 基氧化物与Ga 基氧化物耦合SAPO-34 的双功能催化剂一般可同时适用于CO/CO加氢制低碳烯烃的反应,In基氧化物因易被还原因而多用于CO加氢制低碳烯烃反应中,对于MO(M=Zn、In、Ga)氧化物的设计主要通过掺杂第二元素或掺杂第三助剂构建双活性位点,精准调控表面氧空位含量和H解离能力,实现C—O活化速率和H—H活化速率的匹配,在提高催化活性的同时可抑制烯烃二次加氢。
3 金属氧化物制备方法
纳米金属氧化物常用的制备方法包括共沉淀法、水热法、溶胶凝胶法、溶液燃烧法和MOF 热解法等。不同制备方法对金属氧化物的织构性质、颗粒尺寸、晶型结构、氧空位含量及不同组分间的相互作用都会产生明显的影响,进而影响催化性能。
OX-ZEO催化剂中金属氧化物的制备方法普遍采用共沉淀法,研究发现,沉淀剂的选取对于金属氧化物的物理化学性质及催化性能具有明显的影响。Raveendra 等采用(NH)CO、NaOH、NH·HO 和NaCO作沉淀剂制备了一系列La 掺杂的ZnAlO氧化物复合SAPO-34,发现以NH·HO 作沉淀剂并控制沉淀pH=7 时可获得最佳的催化性能。本文作者课题组Yang 等同样发现,对比采用氢氧化钠、草酸、碳酸钠作沉淀剂制备GaMnO氧化物,采用氨水制备的GaMnO氧化物复合SAPO-34后催化性能最好,在400℃、2.5MPa反应条件下,CO 转化率达到19.5%,低碳烯烃的时空收率为116.1mL/(h·g)。分析认为采用氨水制备的GaMnO氧化物氧空位含量多,Mn 与Ga 之间产生了强的相互作用,有利于提升CO 和H的吸附能力,从而提高催化活性。
采用共沉淀法制备金属氧化物,通过改变前体的焙烧温度可改变所得氧化物的结构和物化性质等。Li 等通过调控焙烧温度(300~1200℃)制备了晶粒尺寸在23~79nm 的ZnO 纳米颗粒,与SAPO-34 复合后,发现CO 转化率和低碳烯烃选择性随ZnO 晶粒尺寸的减少而逐渐增高,证实小晶粒尺寸的ZnO 颗粒有助于提高催化活性,在研究中还发现,小晶粒ZnO 颗粒在反应过程中容易发生团聚长大,导致失活速率较快。Lu 等发现,在煅烧温度300~1200℃范围内可合成7~28nm 的InO颗粒,如图5 所示,焙烧温度越低,InO颗粒尺寸越小,其氧空位含量越高,活化CO和H能力越强,但催化活性和低碳烯烃选择性与颗粒尺寸并非完全呈现正相关性,当颗粒尺寸小于19nm 时,催化剂易烧结,造成催化稳定性变差,颗粒尺寸在19nm 时CO转化率最高,达到14.1%,烃类产物中低碳烯烃选择性达到76.9%,副产物CO选择性为60.9%。
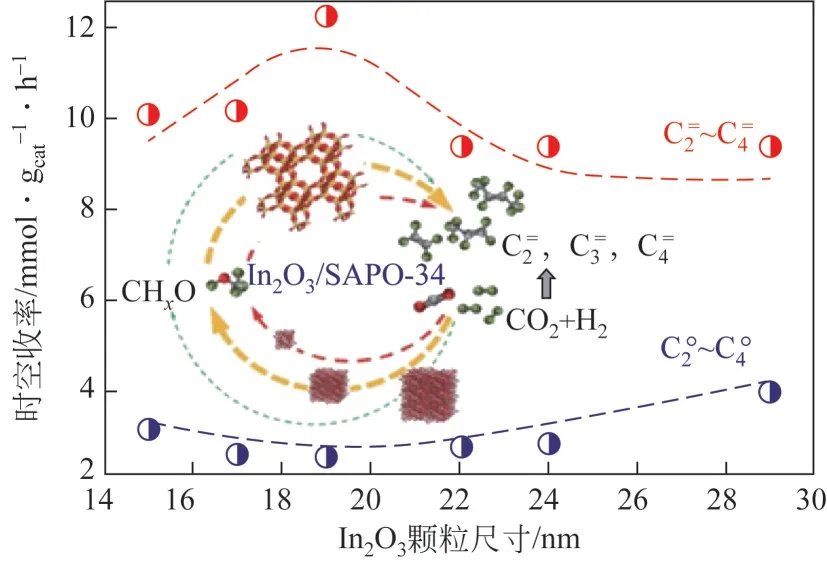
图5 In2O3颗粒尺寸对合成气转化制低碳烯烃反应性能的影响[52]
采用水热法制备金属氧化物,通过改变溶剂的种类可改变ZrO的晶型结构,进而影响催化性能。Liu 等以硝酸氧锆为前体、尿素为沉淀剂,采用水或甲醇为溶剂并通过水热法分别合成了单斜(-)和四方相(-)的ZrO并负载Zn用于STO反应。结果发现,相比于ZnO/-ZrO氧化物,ZnO/-ZrO氧化物可产生更多的表面羟基、路易斯酸及氧空位,有利于反应中间体甲酸根和甲氧基的形成,因而ZnO/-ZrO氧化物复合SAPO-34 后表现出更优的催化性能,在CO 转化率为27.9%时,低碳烯烃选择性高达80.5%,而ZnO/-ZrO+SAPO-34样品的CO 转化率和低碳烯烃选择性分别为16.4%和76.1%。
溶胶凝胶法也是制备金属氧化物有效的方法之一,Luo 等采用溶胶凝胶法制备了ZnCeZrO氧化物,分别采用葡萄糖、柠檬酸、酒石酸、己二酸以及L-丙氨酸作络合剂,发现以葡萄糖做络合剂制备的ZnCeZrO前体经500℃焙烧后产生的氧空位更多,与SAPO-34 复合以后展现出最佳的STO催化性能,在300℃、1.0MPa时,低碳烯烃的时空收率达到0.4mmol/(h·g)。Liu 等以正丁醇锆和正丁醇锌为原料,以正丁醇为溶剂,通过溶胶凝胶法并结合超临界CO干燥技术制备了平均颗粒尺寸仅为4.8nm 的Zn-ZrO氧化物,对比常规以柠檬酸为络合剂制备的颗粒尺寸为8.9nm的Zn-ZrO氧化物,复合SSZ-13后,CO转化率由25%提升至29%。罗耀亚等分别采用共沉淀法、溶胶凝胶法和水热法制备了ZnCeZrO固溶体,其中,以溶胶凝胶法和共沉淀法制备的ZnCeZrO固溶体的形貌为球状堆积,而以水热法得到了棒状结构。通过表征结果发现,以溶胶凝胶法制备的ZnCeZrO固溶体比表面积最大,表面氧空位浓度最高,CO 的吸附活化能力最强,与SAPO-34 复合后的低碳烯烃选择性最高,为79.5%,而以水热法和共沉淀法制备的样品,其低碳烯烃选择性分别为77.0% 和73.1%。
基于MOF 材料热解制备的金属氧化物近年来受到广泛关注。Wang 等以含有部分Zn 取代的UiO-66(Zr-Zn)作为前体,在Ar 气气氛下高温热解制备了具有三维空心结构碳包覆的Zn-Zr 氧化物(HO-ZnZrO@C),如图6 所示。对比常规共沉淀法制备的Zn-Zr 氧化物,采用该方法制备的氧化物颗粒尺寸更小,孔道结构更发达,氧空位含量更多,其丰富的氧空位和较小的颗粒尺寸有助于提供更多的活性位点,发达的孔道结构有助于中间产物的扩散,复合ZSM-5 后由合成气制芳烃,CO 转化率可达35.2%,而常规共沉淀法制备的Zn-Zr 氧化物复合ZSM-5 后CO 转化率仅为16.1%。Liu 等分别采用水热法、UiO-66 热解法、油-水界面合成法、溶胶凝胶法制备了单一的ZrO氧化物并与H-ZSM-5 复合进行合成气芳构化反应,研究表明采用水热法和UiO-66 热解法制备的纳米ZrO颗粒分散性更好,氧空位含量更多,酸性更强,催化活性更高。
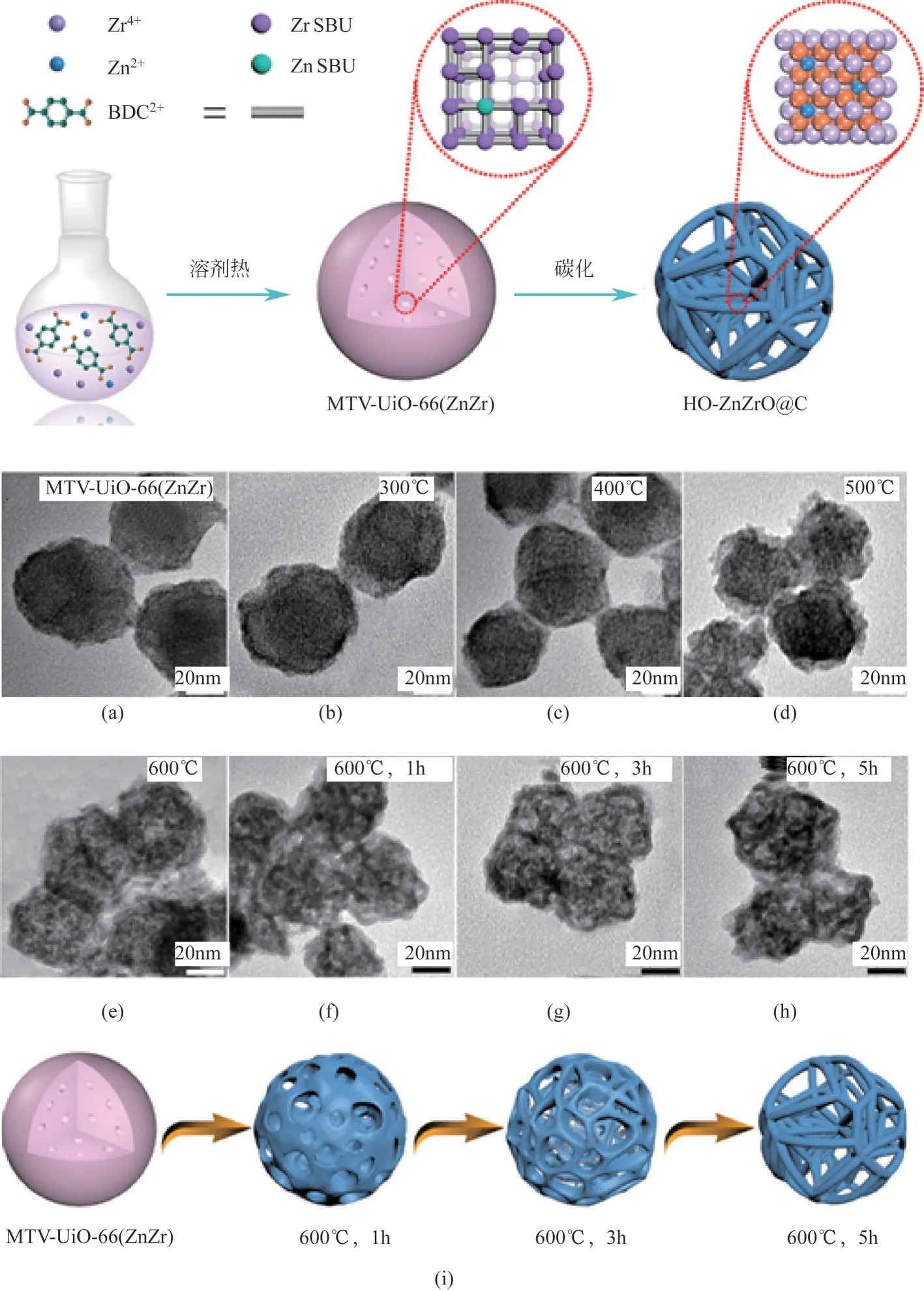
图6 不同温度处理下MTV-UiO-66(ZnZr)到HO-ZnZrO@C结构的演变[56]
4 氧化物和分子筛“亲密度”
目前基于OX-ZEO双功能体系中氧化物和分子筛主要采用物理混合的方式研究二者的“亲密度”,主要包括分层排布、颗粒混合、粉末混合和球磨混合四种装填方式,两者不同的接触方式和接触距离会影响中间产物的形成状态以及传递速率,进而影响催化活性和产物选择性。Cheng 等详细研究了四种装填方式对ZnZrO氧化物复合SAPO-34后催化性能的影响,如图7(a)所示。发现两者分层排布时,分子筛的“拉动效应”被弱化,CO 转化率低,且生成的甲醇中间体会在扩散过程中加氢生成甲烷,当通过颗粒混合、粉末混合、球磨混合装填方式逐渐增加两者“亲密度”时,低碳烯烃选择性逐渐下降,CO 转化率先增加后降低,采用颗粒混合方式低碳烯烃选择性最高,而采用粉末混合时,CO 转化率最高,这说明两者接触并非越紧密越好,保持在纳米尺寸上适中的接触距离分子筛的催化接力效果最为明显。
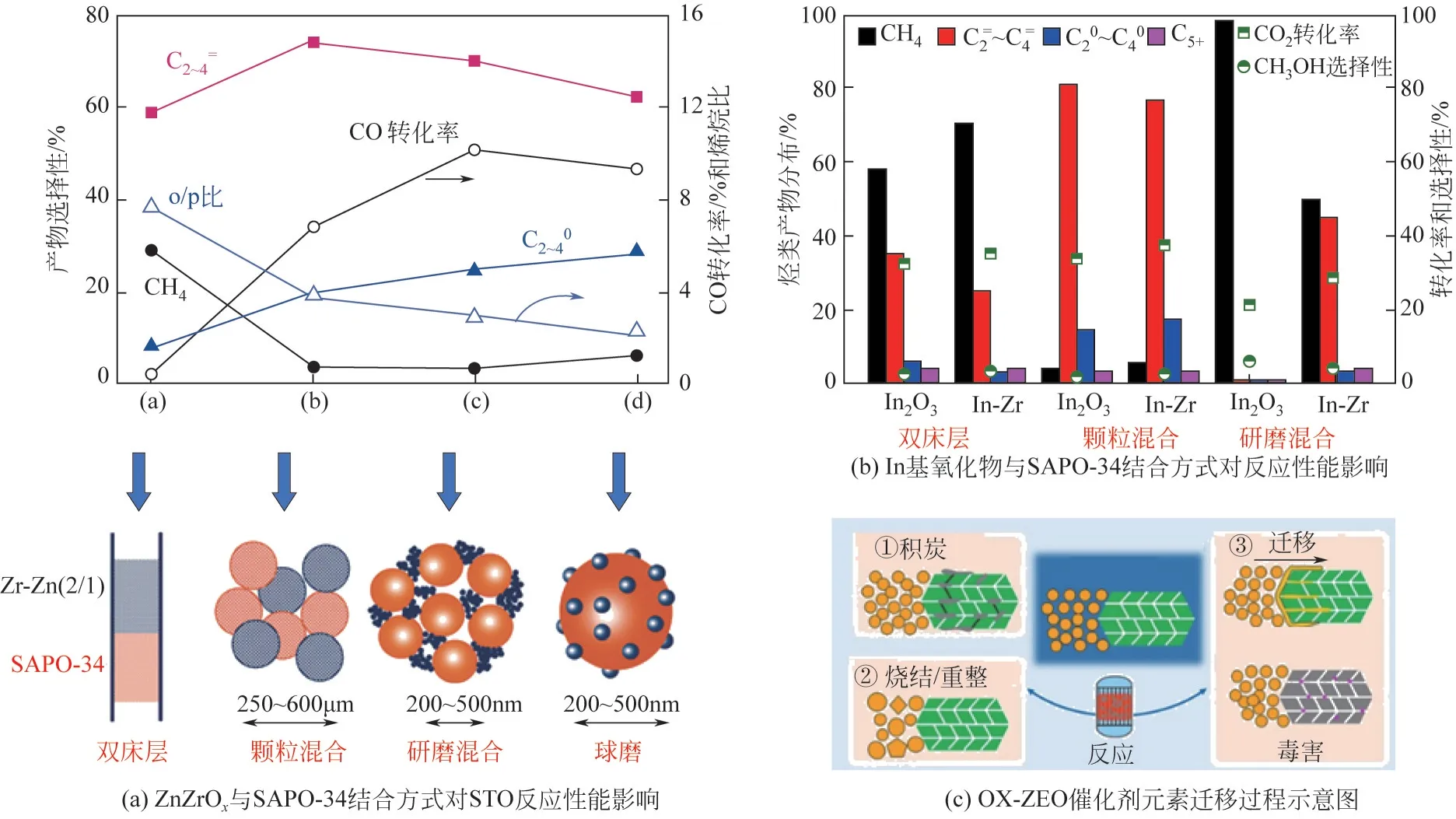
图7 金属氧化物与SAPO-34结合方式对反应性能影响[7,17,60]
采用分层排布装填方式时,尽管分子筛的拉动效果变弱,但是中间产物在分子筛转化过程中生成的水难以再次接触到氧化物表面,有助于抑制WGS 反应,降低CO生成量。例如,Ni 等将ZnAlO氧化物与SAPO-34 分子筛采用分层排布装填方式应用于STO 反应,在CO 转化率为6.9%时,低碳烯烃选择性达到77.0%,副产物CO选择性仅为33.1%。金属氧化物与分子筛接触过于紧密时,可能会发生元素迁移现象,进而影响催化性能。Gao等发现,将InO与SAPO-34采用粉末混合方式应用于CO加氢反应,与颗粒混合方式相比,CO转化率和低碳烯烃选择性明显下降,产物甲烷选择性达到95%,如图7(b)所示。主要原因是InO与SAPO-34 接触过于紧密时,反应过程中会导致In 物种迁移至SAPO-34 表面并与分子筛质子进行离子交换,降低了分子筛强酸位点,导致催化剂的快速失活和过度加氢。Ding 等在ZnCrO/SAPO-34 上也发现类似现象,ZnCrO与SAPO-34 接触距离过近时,Zn 物种会迁移至SAPO-34 的Brønsted酸位点形成Zn-OH,导致了烯烃的过度加氢。Wang等详细对比了单一InO、ZnO、CrO、ZrO耦合ZSM-5 后元素的迁移能力,发现InO和ZnO在反应过程中元素迁移能力最强,ZrO迁移能力最弱,并推测元素的迁移能力可能与其还原能力有关,还原能力越强的氧化物越容易发生金属离子的迁移。如图7(c)所示,元素迁移现象通常发生在催化剂反应过程中,元素迁移会毒害分子筛的Brønsted 酸位点,导致催化剂活性和稳定性均降低。
因此,基于InO和ZnO 高的可还原性,目前报道的In 基和Zn 基的双功能催化剂普遍采用颗粒混合的结合方式以减弱金属离子迁移,提高催化剂稳定性。然而,本文作者课题组Zhang 等发现,对于Ga 基双功能催化剂,采用粉末混合方式对比颗粒混合方式,其CO 转化率和低碳烯烃选择性更高,表现出“越近,越好”的特点,说明Ga 基氧化物中Ga物种可能更稳定,且由于GaO难以被还原而没有出现Ga物种迁移现象。
最近,Tan 等发现核壳结构的OX-ZEO 催化剂表现出更优的催化性能,该研究以Zn-Cr氧化物为核,以SAPO-34 为壳制备出了Zn-Cr@SAPO-34催化剂,如图8 所示。对比传统的物理混合方式,CO 转化率和低碳烯烃选择性更高,CO选择性更低,仅为36%。他们将活性提高的原因归因于这种核壳结构导致Zr-Cr 氧化物与SAPO 接触更紧密,缩短了中间产物到SAPO的扩散路径。另外,研究还发现,SAPO-34 分子筛对水的吸附能力更强,因此以SAPO-34 作为壳层反应过程中生成的水会优先吸附到SAPO-34 上,一定程度上减弱了核层Zn-Cr 氧化物对水的吸附,抑制了WGS 反应发生,减少了CO的生成。
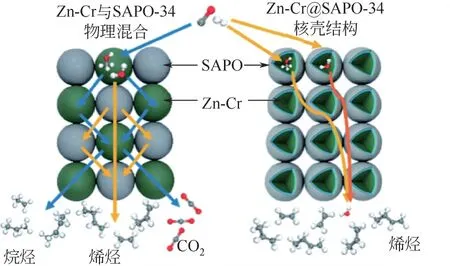
图8 物理混合的Zn-Cr/SAPO-34与核壳结构Zn-Cr@SAPO-34催化剂不同的传质过程的对比[61]
5 催化反应机理
对于OX-ZEO双功能催化剂,尽管研究人员对其反应机理进行了深入的研究,但对反应中间物种的认知以及中间物种的形成路径仍存在争议,根据金属氧化物对CO和H的活化方式不同,目前主要提出了两种不同的反应机理:烯酮中间体(CHCO) 或甲醇/二甲醚中间体 (CHOH/CHOCH)路径。
5.1 烯酮中间体
烯酮中间体路径最早由Jiao 等提出,他们以ZnCrO/SAPO-34 为研究对象,在ZnCrO氧化物表面通入合成气,通过同步辐射真空紫外光电离质谱(SVUV-PIMS)表征手段观察到了烯酮物种,如图9(a)所示,且实验证实以烯酮为原料合成低碳烯烃的催化稳定性远高于以甲醇为原料,这与OX-ZEO反应过程的高稳定性相一致,因此认为烯酮是连接ZnCrO氧化物与SAPO-34 分子筛的中间体。烯酮中间体完整的生成路径如图9(b)所示,CO 在部分还原的ZnCrO氧化物产生的氧空位处吸附解离,生成表面的C物种和CO。C与吸附的H结合生成CH,CH*从金属氧化物表面脱附,与气相中的CO 结合生成CHCO 中间体,然后CHCO 中间体扩散到SAPO-34分子筛的孔道中进而生成低碳烃类。Li 等通过高灵敏度低能离子散射光谱(HSLEIS)、-FTIR、XPS、TPD 等手段证实ZnCrO氧化物的预还原过程对于CO 的直接解离起到至关重要的作用,还原温度越高,ZnCrO氧化物的还原能力越强,进而导致对CO 的解离能力越强,Cr 的存在增强了生成C物种和含氧碳物种的能力。除ZnCrO外,他们认为在部分还原的MnO上合成气转化同样遵循烯酮反应路径。
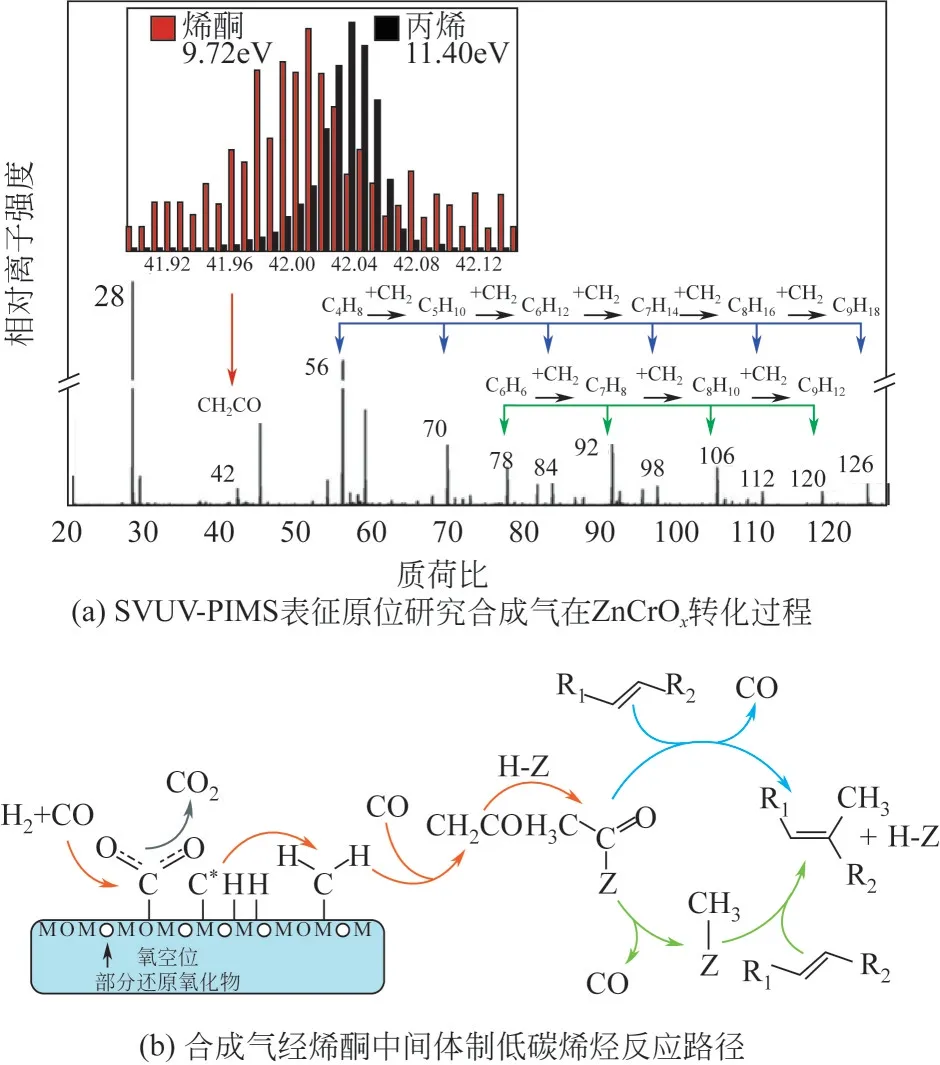
图9 合成气经烯酮中间体合成低碳烯烃反应机理[5,13,63]
5.2 甲醇/二甲醚中间体
近年来,随着对OX-ZEO双功能催化体系研究的加深,该反应路径普遍被认为是甲醇合成反应与甲醇制烯烃反应的耦合,因而以甲醇/二甲醚为中间产物的观点更为广泛接受。Ni等将合成二甲醚的催化剂ZnAlO与MTO 反应催化剂SAPO-34 分层装填,实现了合成气到低碳烯烃的高选择性转化,因此认为OX-ZEO 反应过程为合成气到二甲醚(STD)和MTO 反应的串联,二甲醚为反应的中间体。Liu等通过原位红外表征在Zn-ZrO氧化物上检测到了HCOO和CHO的红外信号峰,如图10(a)所示。他们认为CO 与Zn-ZrO氧化物表面羟基反应生成HCOO,HCOO与Zn 位点解离的H反应并持续加氢生成CHO,进而转化为甲醇和二甲醚反应中间体,反应机理如图10(b)所示。借助原位红外表征手段,除Zn-ZrO氧化物外,其他金属氧化物 如 ZnAlO、 ZnGaO、 CeZnZrO、ZnMnO、CrMnGaO、GaZrO等吸附CO+H后同样可观察到HCOO和CHO信号。此外,有研究认为,形成甲醇过程的中间含氧物种(CHO)也有可能是反应的中间体,Li等通过原位红外表征以及化学捕集-质谱法研究了CO加氢在ZnZrO/SPAO-34 催化剂表面的转化过程,证实CHO、CHO、CHOH 均有可能从ZnZrO氧化物表面脱附进入到SAPO-34孔道转化成低碳烯烃。
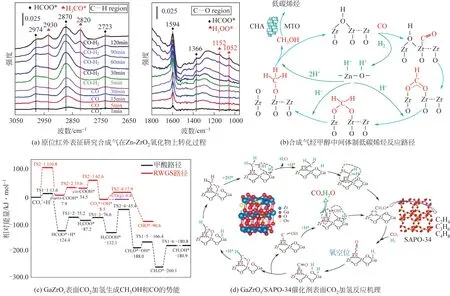
图10 COx加氢经甲醇/二甲醚中间体合成低碳烯烃反应机理[10,20]
值得注意的是,基于OX-ZEO 双功能催化剂,无论是合成气转化还是CO加氢反应,虽然通过红外表征证实反应中间产物是甲醇等含氧物种,但是甲醇的形成路径不尽相同。在STO反应中,研究者普遍认为金属氧化物表面羟基参与了中间体的形成。对于CO加氢反应,含氧中间体的生成理论上存在两种反应路径:一是CO加氢先经RGWS反应生成CO,然后CO 加氢生成中间体;二是CO直接在氧化物缺陷位点活化并通过加氢生成反应中间体,后者在OX-ZEO 催化剂上更为认可和接受。本文作者课题组Zhang 等以O-GaZrO(101)为模型,通过DFT理论计算研究了CO加氢分别经甲酸路径和RWGS路径形成CHO的反应势能,如图10(c)所示。计算结果表明,CO经RWGS反应生成后更容易从GaZrO氧化物表面脱附出来形成气相CO,而非继续加氢形成含氧中间体,CHO经HCOO路径转化在能量上则更为有利。其完整的加氢路径如图10(d)所示,CO在Zr—O—Zr位点吸附活化,同时H在Ga—O 位点吸附解离,生成Ga—H和O—H,活化的CO既可以与Ga—H反应并逐步加氢生成HCOO和CHO反应中间体,又可以与O—H反应生成COOH,并进一步解离形成CO。
5.3 氧空位作用
金属氧化物在焙烧或还原过程中容易导致部分晶格氧脱除形成氧空位,基于烯酮中间体机理,研究者认为被束缚在金属氧化物氧空位中的未成对电子可以弱化C—O键,从而使得CO 发生歧化反应,解离生成C物种和CO物种,CO解离脱附生成副产物CO,C物种参与反应中间体的形成。Jiao等采用DFT理论计算表明,ZnCrO(111)表面是可还原的,还原过程中产生大量氧空位,CO 吸附能随表面氧空位含量的增多而降低。而对于甲醇中间体路径,研究者普遍认为氧空位的存在有助于CO的非解离活化,Wang 等研究表明,CO 转化率随ZnCeZrO氧化物表面氧空位的含量线性增加,说明氧空位的存在有助于CO转化率(图11)。Liu等发现Zn-Ga氧化物上CO转化生成甲醇的速率与氧空位浓度一致,说明了氧空位在CO活化过程中的重要作用。Zhou 等和Zhang 等分别在ZnO-ZrO和GaZrO氧化物上也发现类似的规律,即CO转化率与这些氧化物氧空位的浓度呈现正相关性。
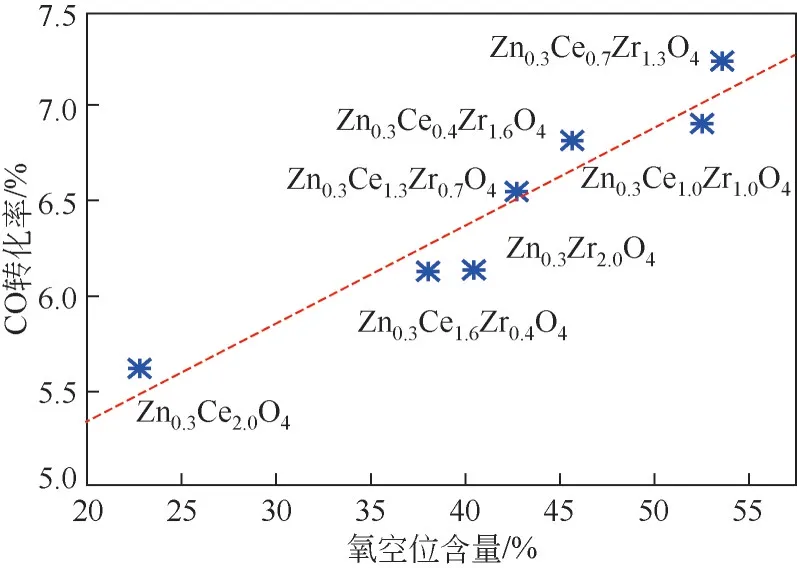
图11 金属氧化物表面氧空位含量和CO转化率关系[26]
尽管大多数研究者认可氧空位对CO的积极作用,然而部分研究认为氧空位对CO 加氢生成甲醇的过程是不利的。Su 等通过对Zr-InO氧化物的理论计算发现,CO与主要晶格氧反应生成OCO物种,进而加氢生成甲醇,而在氧空位处甲醇会发生C—O 键的断裂生成甲烷,即甲醇中间体是在Zr-InO氧化物完美的晶面上生成的,缺陷位是生成副产物甲烷的活性位点。Fu 等以ZnO 为研究对象,建立了合成气到甲醇、烯酮、甲烷转化的完整反应网络,通过DFT 理论计算,采用反应相图系统研究了合成气在ZnO 表面的转化,计算结果表明,ZnO表面的氧空位含量是影响产物选择性的关键因素,氧空位含量较低时,甲醇更容易生成,随着氧空位含量的增加,产物由甲醇向烯酮转化,氧空位含量过高时产物以甲烷为主。
5.4 降低副产物COx策略

在CO加氢反应中,由RWGS 反应产生的CO为主要副产物,RWGS与甲醇合成反应相竞争,热力学性质决定了高温更有利于RWGS反应,因此通过降低反应温度可抑制RWGS 反应减少CO 的形成,但与此同时催化活性也会降低。此外,改变反应工艺条件如提高H/CO比、通入CO 原料气也可抑制RWGS反应。需要强调的是,除了反应条件因素影响外,金属氧化物本身的性质同样决定着其RWGS反应的难易程度,研究者普遍认为甲醇等含氧中间产物的形成速率依赖于金属氧化物表面氧空位的含量,因此可对氧化物活性组分通过助剂修饰,改变制备方法等手段提高氧空位含量,使反应更有利于往合成甲醇方向转化,进而抑制RWGS反应以减少CO的生成。
6 结语
烯烃原料多元化是我国未来烯烃产业的重点发展方向,通过FTTO 或OX-ZEO 路线可将CO加氢一步转化成低碳烯烃,前者虽然可以获得较高的CO转化率,但由于反应服从聚合机理导致产物分布宽,产品组成复杂,工业化进程缓慢,最近的研究采用疏水改性提高催化剂的烯烃收率,但产物分布仍遵循ASF 分布规律,低碳烯烃选择性较低;采用OX-ZEO催化剂产物脱离费托反应路线ASF分布限制,低碳烯烃选择性高达70%~80%,具有较好的工业应用前景。但该路径仍然面临诸多问题,如:①产物低碳烯烃在高温、H气氛下的过度氢化;②催化活性仍较低,副产物选择性高,造成原子利用率低。因此在维持高选择性的基础上进一步提高催化活性仍然是该研究领域面临的最大挑战。开发高效的OX-ZEO 双功能催化剂,尤其设计出H解离能力适中的高温甲醇或类甲醇合成催化剂,使之与MTO 反应温度区间匹配,实现双功能催化剂两组分之间的“热耦合效应”,同时避免烯烃产物的过度氢化,是制备高活性和高选择性双功能催化剂的关键。目前,氧化物研究的热点主要针对Zn 基、In 基和Ga 基氧化物改性,通过掺杂第二元素、助剂、改变催化剂制备方法等手段来调控氧化物表面氧空位含量和H解离能力,促进CO/H的吸附活化,改善催化性能。
尽管OX-ZEO双功能催化剂在调控低碳烯烃选择性上优势明显,但由于较低的单程CO转化率和较高的副产物选择性,导致其低碳烯烃收率仍然较低(<30%),在维持高选择性前提下进一步改善CO转化率以及抑制副反应发生,是OX-ZEO 催化体系亟需解决的关键问题。对比Zn 基和In 基氧化物,Ga 基固溶体氧化物耦合SAPO-34 在催化活性和低碳烯烃选择性上表现出更优异的催化性能,如何精心地设计Ga 基双功能催化剂,使其在高空速下的催化活性与FTO 路径相竞争值得期待。研究者普遍认为金属表面氧空位是CO活化的活性位点,可进一步通过元素掺杂以及助剂修饰来提高金属氧化物表面氧空位含量,进而提高催化活性;除元素修饰外,氧化物的制备方法同样影响氧空位含量,通过采取合适的制备方法制备出大比表面积、粒径尺寸小的氧化物有利于暴露出更多的氧空位,进一步改善催化性能,然而小晶粒的氧化物抗烧结能力和稳定性差,如何实现氧化物小晶粒和高稳定性的兼顾是未来的一个研究方向;金属氧化物与分子筛“亲密度”是影响催化性能的关键因素,不同的氧化物类型与分子筛最优的接触距离不尽相同,对于元素易迁移的氧化物组分,除调控两者接触距离外,仍需对氧化物通过助剂修饰等方法来提高氧化物结构稳定性,抑制氧化物中元素的迁移;此外,高温条件下STO 反应中伴随的副反应WGS 反应或CO加氢过程中伴随的RWGS 反应不可避免,导致C 原子的利用率较低,未来仍需对催化反应机理及动力学进行深入研究,通过助剂修饰、催化剂结构设计和工艺条件优化等方面来提高甲醇等中间产物的生成速率,抑制副反应的发生,提高C 原子利用率。如针对STO 反应,可将费托反应中核层碳化铁活性相与壳层疏水基团的高效协同性扩展到OX-ZEO催化剂的设计,通过对氧化物进行疏水改性,抑制反应产生的HO 在氧化物上的吸附和停留,进而抑制WGS 反应,降低CO排放量,与此同时,疏水壳层的厚度对中间体扩散速率影响、高温反应下疏水基团稳定性等关键问题值得考虑和深入研究。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
化工进展(2022年5期)2022-05-26
科学家(2022年4期)2022-05-10
当代化工(2020年6期)2020-08-24
世界家苑(2020年4期)2020-06-30
山东工业技术(2018年9期)2018-05-26
股市动态分析(2015年12期)2015-09-10
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
城市建设理论研究(2012年35期)2012-04-23
