
铀酰-(6′-(1H-吡唑-1-硫代羰基)-[2,2′-联吡啶]-6-基)(5-甲氧基-1H-吡唑-1-基)甲硫酮对手性丙溴磷络合分离的理论研究
2022-08-22 03:29:54刘林峰聂长明
南华大学学报(自然科学版) 2022年3期
刘林峰,聂长明
(南华大学 化学化工学院,湖南 衡阳 421001)
0 引 言

有机磷手性农药在过去数十年里被广泛应用于害虫防治,灭虫机理是使生物体内的乙酰胆碱酯酶磷酸化,从而失去分解乙酰胆碱的能力[9-10]。图1中的R/S-丙溴磷(R/S-profenofos,R/S-PFFs)是一种常见的有机磷手性农药。手性丙溴磷农药的R/S构型对人体的生物毒性大为不同,识别和分离手性丙溴磷农药对环境保护和食品安全有着重要作用[11]。
目前含不对称的硫代酰胺配体的铀酰络合物及其对手性丙溴磷农药的分子识别和分离未见报道。受上述启发本文从理论上设计了一种新颖的硫代酰胺配体(6′-(1H-吡唑-1-硫代羰基)-(2,2′-联吡啶)-6-基)(5-甲氧基-1H-吡唑-1-基)甲硫酮(6′-(1H-pyrazole-1-carbonothioyl)-[2,2′-bipyridin]-6-yl)(5-methoxy-1H-pyrazol-1-yl)methanethione(PMMT),将配体PMMT与铀酰配位络合构建Uranyl-PMMT受体(如图1),再用受体识别和分离手性R/S-丙溴磷对映异构体,从理论上探讨了这种含不对称硫代酰胺配体对丙溴磷对映异构体络合能力与选择性,揭示了受体对R/S-PFFs对映异构体识别和分离的本质。
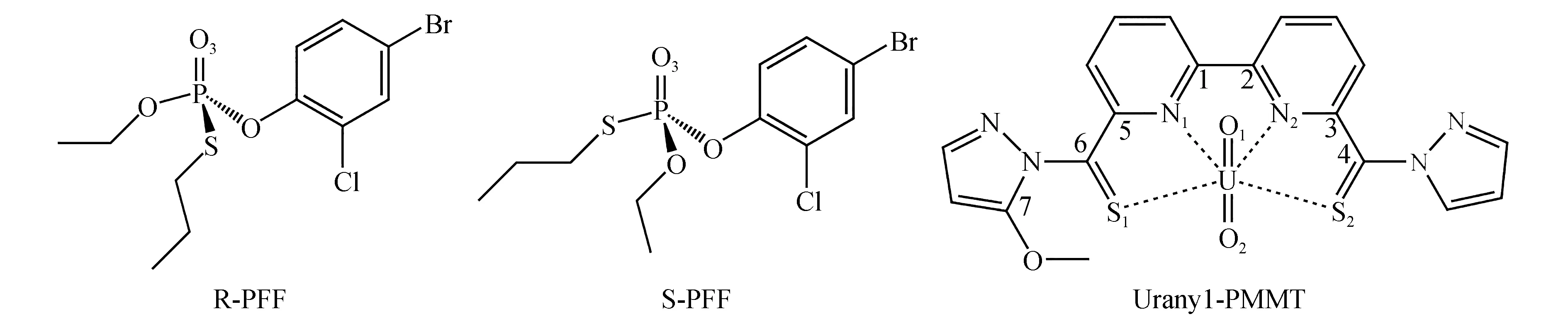
图1 R/S-丙溴磷和Uranyl-PMMT的化学结构式
1 计算方法
在gaussian 16软件[12]中使用BP86[13]泛函对配体及其络合物进行了优化。考虑到相对论效应对锕系元素d轨道和f轨道的间接影响十分显著,用小核标量相对论有效核势(scalar-relativistic effective core potentials,RECPs,60 core electrons)来描述铀原子的相对论效应,并用相应的ECP60MWB[14]基组来描述铀原子,对于其他轻原子则采用def2-SVP基组来描述。
本文计算了配体在气相中的表面静电势(electrostatic potential,ESP),并在此基础上分析了受体-客体配合物的几何结构。通过Multiwfn[15]与VMD软件[16]的结合,获取了受体-客体配合物的定域化轨道指示函数图(localized orbital locator,LOL)、红外光谱图和前线分子轨道。然后在Multiwfn软件中使用分子中的原子量子理论(quantum theory of atoms in molecules,QTAIM)对受体-客体配合物进行了电子密度拓扑分析。在BP86/def2-TZVP/RECP水平上计算了所有化合物的气相单点能,再采用SMD[17]溶剂模型来评估其在水和有机溶剂中的溶剂效应。
2 结果与讨论
2.1 配体的表面静电势
静电势被广泛使用于考察分子间静电相互作用和预测反应位点[18-19]。图2展示了PMMT配体的表面静电势图,红色区域具有亲电活性,静电势为正值,带负电荷的微粒容易靠近。蓝色区域具有亲核活性,静电势为负值,与带正电荷的微粒有较强的相互作用。由图2可知所有配体的静电势负值主要出现在S和N原子上,说明这些位点有较强与铀酰离子配位的能力。配体PMMT最负的静电势值位于其空腔内,说明PMMT的硫和氮原子电子密度大,给电子能力强,因此,铀酰离子更倾向于填充到配体的空腔中。
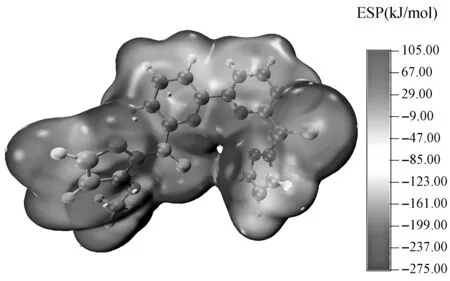
图2 PMMT的表面静电势图
2.2 受体及其配合物的结构
在BP86/def2-SVP/RECP级别上优化了受体及其配合物的结构,所有优化后的配合物力与位移均收敛且没有虚频,说明配合物的结构稳定是局部势能面的能量最低点。配体PMMT含有多个供体原子,只有其连二吡啶上的两个吡啶氮和两个硫代甲硫酰基上的硫原子能与铀酰配位形成稳定的受体,由于铀酰最稳定的配位数是5,受体还有一个配位活性点可以与客体的配位原子配位从而对丙溴磷对映异构体进行识别分离。手性客体R/S-丙溴磷中的磷酰基氧(客体含有6个供电子体,只有磷酰基氧与受体配位收敛)可以与受体Uranyl-PMMT发生络合作用而形成5配位的配合物R/S-PFF-Uranyl-PMMTs,图3展示了受体及其配合物名字。
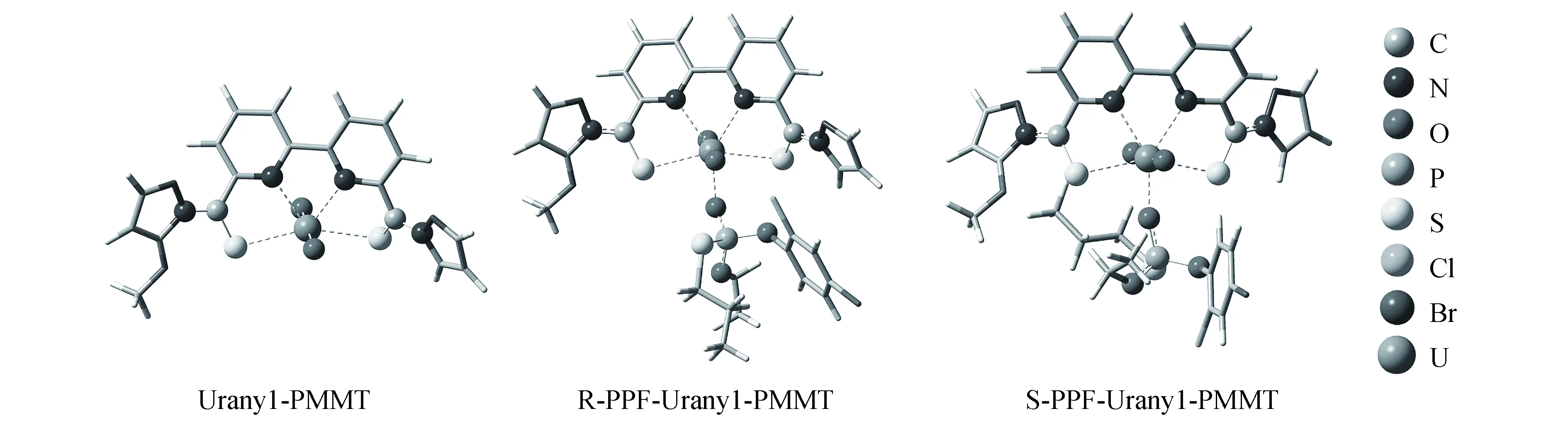
图3 BP86/def2-SVP/RECP水平下优化的受体和受体-客体络合物的结构模型
表1给出了受体与受体-客体配合物的键长、键角和二面角的计算结果。结果表明,配合物中U—S、U—N、U—O1和U—O2的键长基本都大于配位前受体的键长。可能是由于受体与客体配位形成了U—O3配位键,降低了其他配位键的强度。同时由于硫的有效原子半径比较大,U—N键和U—O键明显短于U—S键。总体上来说,S型配合物的键长比R型的键长要短,表明Uranyl-PMMT受体对客体S-丙溴磷有更强的配位能力。铀酰离子的键角是180°,但在R-型和S-型的配合物中O1—U—O2的平均键角分别为176.73°和175.93°,说明铀酰离子键角发生了轻微扭曲。而所有配合物中的S1—U—S2的键角明显大于配位前,表明受体与客体配位后空间结构发生了明显变化。对比二面角C1—N1—C5—C6、N1—C5—C6—S1、S1—U—N2—N1的结果,发现S型络合物的二面角比R型络合物的偏转角度更大一些,这可能导致受体与S型丙溴磷络合时的空间位阻比与R-PFF络合时更小,所以S型配位络合物的键长比R型的键长要短。
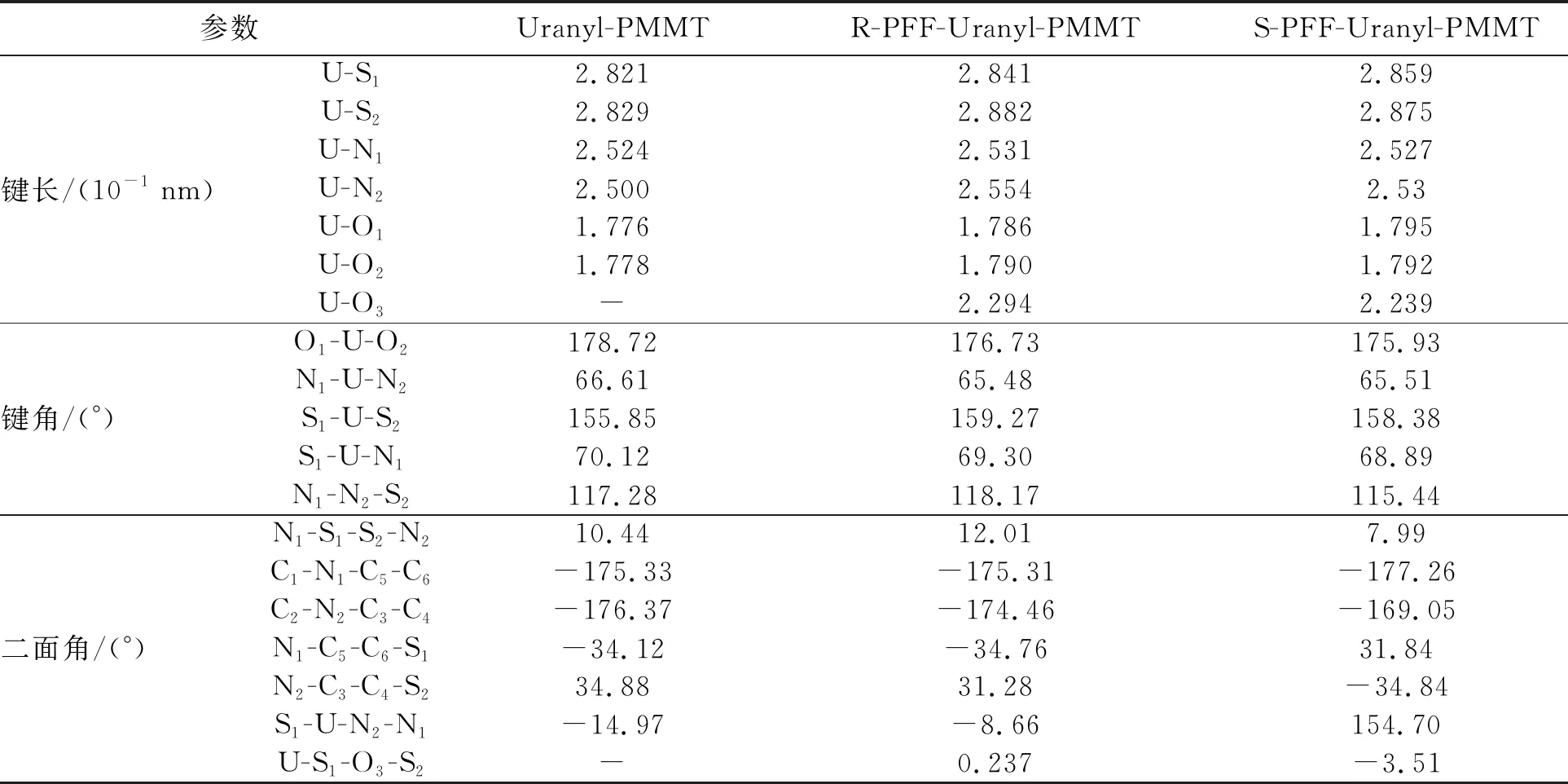
表1 受体及其配合物的计算几何结构参数
2.3 Mayer键级、分子中原子量子理论分析
Mayer键级(mayer bond order,MBO)是衡量化学键相对强弱的参数,键级越大,键越稳定。从物理上可以理解为原子间共享的电子对数,在BP86/BS-I/RECP级别上计算了所有配合物的Mayer键级,部分关键数据如表2所示。由表2可以看出U—N1和U—N2的MBO值在0.38~0.40之间,说明他们的共价性较低,并且在R和S型配合物中基本保持不变,表明N1,N2两个配位原子对手性R/S-PFF的分离影响不大。U—S1的MBO值比U—S2的MBO值要大,表明U—S1键的共价组分更高。客体的P—O3键MBO值都明显减小,说明受体对客体有明显的配位和识别作用,且S型络合物的P—O3键减小得稍多一些,表明受体对S型丙溴磷的络合识别能力更强。U—O3配位键的MBO值较大在0.65左右,共价组分较高,且PMMT的S型络合物MBO数值比R型络合物要大,说明Uranyl-PMMT能有效地区分丙溴磷对映异构体,且Uranyl-PMMT对S-丙溴磷异构体的配位能力更强。
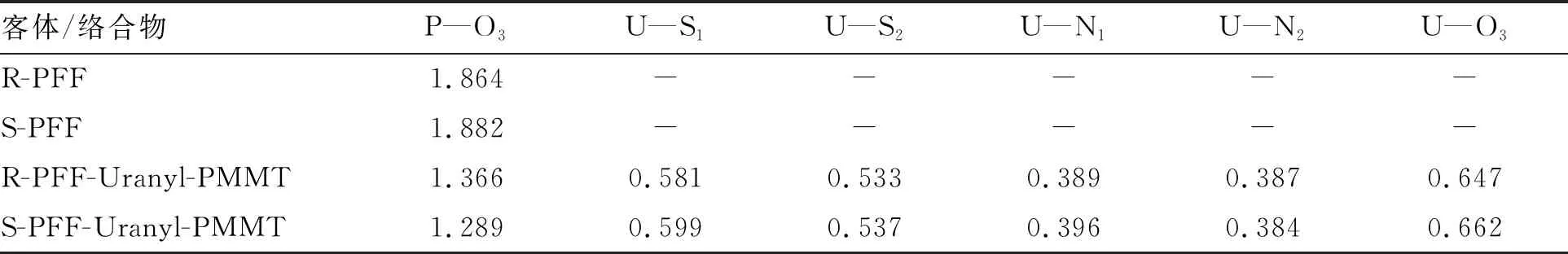
表2 计算的客体及其络合物的部分Mayer键级
为了评估受体-客体络合物五个配位键的共价性,在Multiwfn中使用QTAIM方法对配位键进行了电子密度拓扑分析。AIM(atoms in molecule)理论中BCP(bond critical,键临界点)上的电子密度(ρ)和它的拉普拉斯(2ρ)可以用来考察相应化学键的性质和强度。一般情况下,BCP上的ρ值>0.2 a.u.且2ρ<0时被认为是典型的共价键,当BCP上的ρ值<0.1 a.u.且2ρ>0则是离子键。受体-客体配合物U—S,U—N和U—O3键BCP处的电子密度(ρ)和拉普拉斯(2ρ)列于表3。ρ和2ρ的值表明U—S,U—N和U—O3键是弱的共价相互作用。受体的U—S键和U—N键在与客体配位后共价性明显变小。受体-客体络合物的U—S1键BCP处的ρ值比U—S2键处的ρ值略大,表明PMMT的U—S1键具有更大的共价性。此外,S型的络合物ρ值比R型的ρ值大,说明S型络合物的共价性更高,这与Mayer键级的结果一致。
表3 受体-客体络合物配位键BCPs处的计算拓扑性质/分别代表ρ和2ρ的值
Table 3 Calculated topological properties at BCPs of receptor-guest complexes coordination bonds (单位:a.u.)
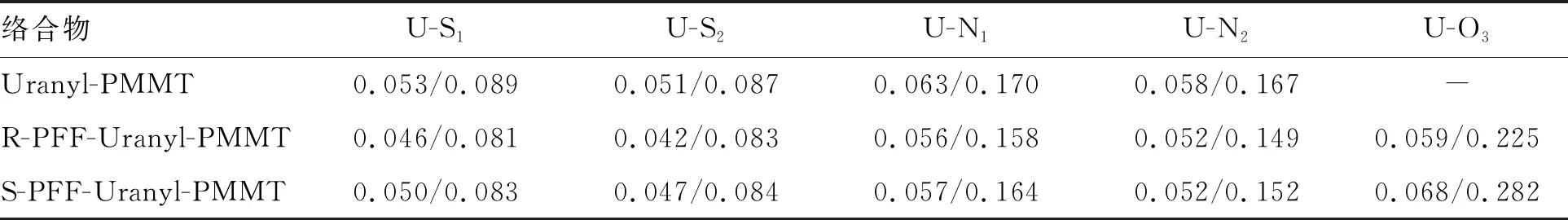
表3 受体-客体络合物配位键BCPs处的计算拓扑性质/分别代表ρ和2ρ的值
络合物U-S1U-S2U-N1U-N2U-O3Uranyl-PMMT0.053/0.0890.051/0.0870.063/0.1700.058/0.167-R-PFF-Uranyl-PMMT0.046/0.0810.042/0.0830.056/0.1580.052/0.1490.059/0.225S-PFF-Uranyl-PMMT0.050/0.0830.047/0.0840.057/0.1640.052/0.1520.068/0.282
2.4 红外光谱分析
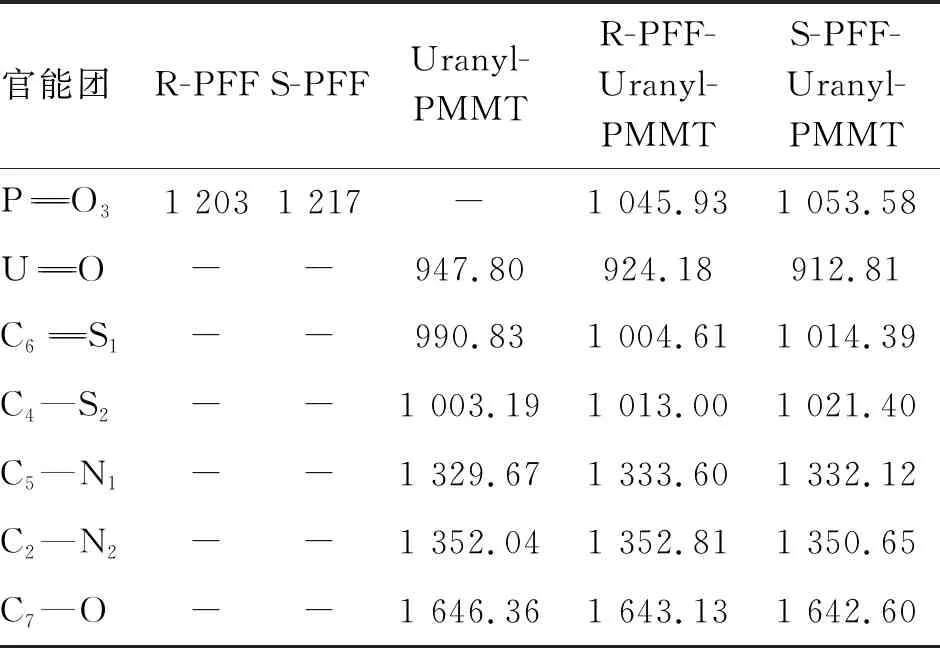
表4 客体,受体以及受体-客体络合物的主要红外数据
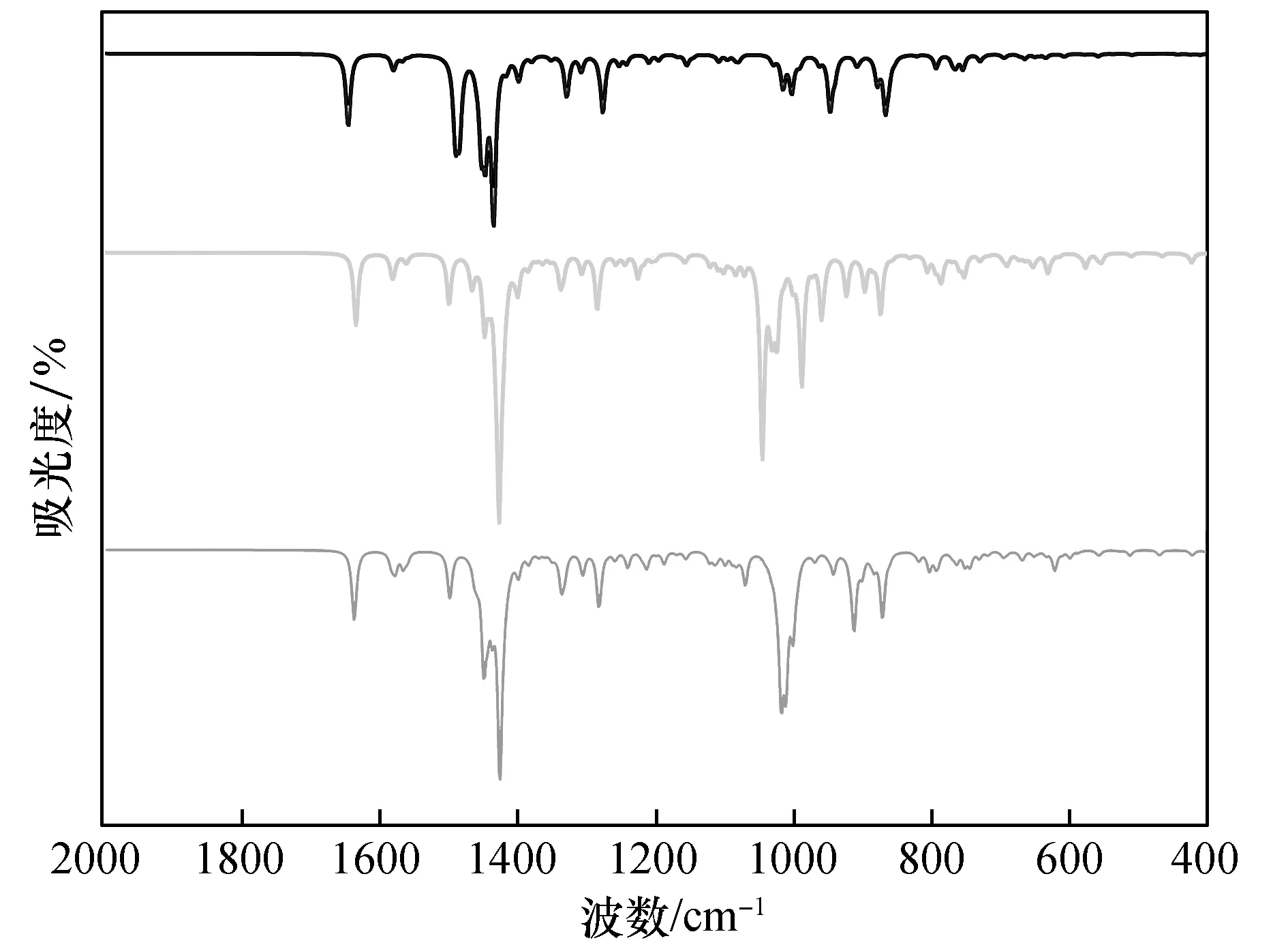
图4 受体及其络合物的红外光谱图
2.5 轨道分析
前线轨道理论是由日本化学家福本谦一教授提出的一种化学理论,其认为最高占据分子轨道(highest occupied molecular orbital,HOMO)和最低未占据分子轨道(lowest unoccupied molecular orbital,LUMO)是决定一个体系发生化学反应的关键。最高占据轨道上的电子所受束缚最小,最容易发生跃迁;最低未占据轨道的能量最低,最容易接纳电子。HOMO-LUMO能隙越大,说明电子基态稳定性越强。通过计算得到了受体和配合物的前线分子轨道能量ELUMO和EHOMO以及能隙ΔEL-H(ELUMO-EHOMO)如图5所示,Uranyl-PMMT受体的能隙ΔEL-H分别为2.65 eV,他们与R/S-丙溴磷配位后能隙ΔEL-H变大了,揭示了Uranyl-PMMT与客体配位后基态电子稳定性得到提升。总体来说,S-PFF-Uranyl-PMMT的ΔEL-H比R-PFF-Uranyl-PMMT大,说明受体更容易与S型丙溴磷形成稳定配合物。
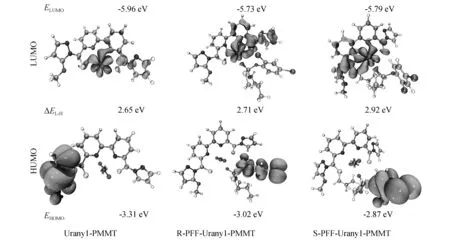
图5 受体及其配合物的前线分子轨道和轨道能量
定域化轨道函数(LOL)可以显示分子不同部分的动能密度差异。原子周围的等值面数额越高,其电子局定域化越强。图6显示了受体-客体络合物的定域化轨道函数图,可以明显看出受体-客体络合物配位键中间的成键区域。U原子与五个配位原子之间的高定域性区域非常弱,被认为具有弱共价性。U和S1之间的LOL值大于U和S2之间的LOL值,这表明受体与客体配位时S1原子起着更大的作用。观察图6可知,S1、S2、N1、N2和O3原子与U原子形成了配位键,其中S型络合物U—S键的LOL值比R型络合物的要大,说明受体与S型丙溴磷的配位更加稳定。

图6 受体-客体络合物的定域化轨道函数图
2.6 配合物的热力学性质
在此次研究中使用化学方程式:受体+R/S-PFFs→受体-R/S-PFFs来计算配体的热力学性质[20]。表5给出了R-PFF-Uranyl-PMMT和S-PFF-Uranyl-PMMT受体-客体络合物在真空、水和有机溶剂中的Gibbs自由能变。表中的数据由以下公式计算得出:
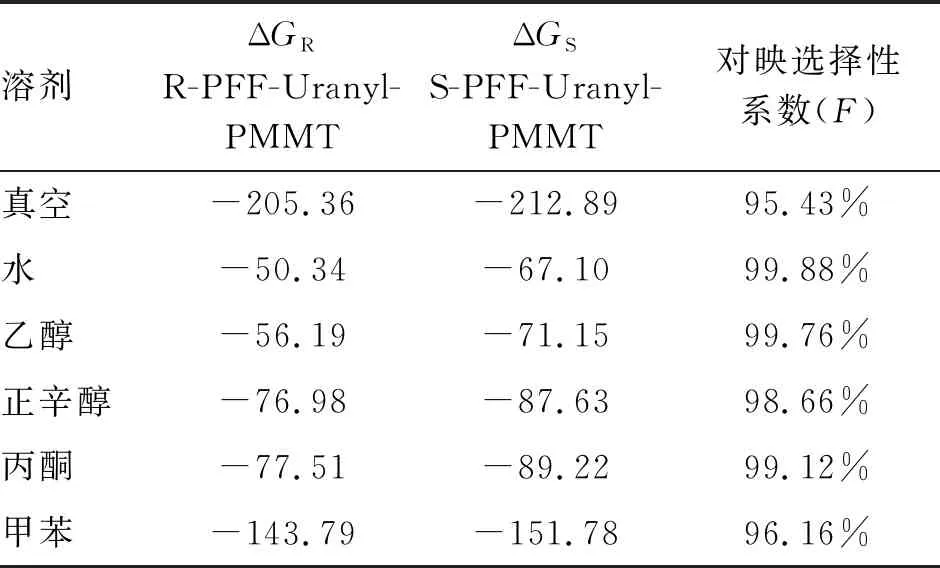
表5 受体-客体络合物的吉布斯自由能变ΔG(kJ/mol)及其对映选择性系数
G=Ggas+Gsol+Gc
(1)
ΔG=(GAB-GA-GB)×627.51
(2)
式(1)中,Ggas为物质在1.01×102kPa气相下的自由能,Gsol是物质的溶解自由能,Gc是物质在标准状况下由1.01×102kPa下的浓度向1 mol/L浓度转变时对应的自由能变(7.917 kJ/mol)。G表示为受体、客体和受体-客体络合物在溶剂环境下标况(298.15 K、1 mol/L)时的自由能。
式(2)中,GAB,GA和GB分别为配合物,受体和R/S-丙溴磷的自由能,ΔG表示整个反应体系的吉布斯自由能变,单位为kJ/mol。627.51为单位换算时需要乘的数值。
R-PFF-Uranyl-PMMT和S-PFF-Uranyl-PMMT络合物的ΔG值均为负值,表明所有受体对客体均具有显著的络合能力。并且同一受体在不同溶剂中的ΔG也明显不同,受体-客体络合物在甲苯中的ΔG最负,说明受体在甲苯中与R/S-丙溴磷的结合能力最好。受体-客体络合物在真空中的ΔG最负,表明受体与客体在真空中的结合能最大,反应最容易发生。通过对比所有络合物的ΔG发现,S-PFF-Uranyl-PMMT的ΔG总体上比R-PFF-Uranyl-PMMT的ΔG要稍微大一些,揭示了Uranyl-PMMT与S型丙溴磷的结合能更强。
在此次研究的分离体系中,根据不同构型络合物之间的吉布斯自由能变可以算出受体对手性R/S-丙溴磷的对映选择性系数。计算公式如下:
(3)
(4)
ΔΔGS/R表示S型络合物和R型络合物之间的吉布斯自由能差;KR和KS分别表示Uranyl-PMMT对R-PFF和S-PFF分子的识别系数;R为气体摩尔常数,T=298.15 K。
由表5可知,所有F值均大于90%,表明受体对R/S-PFFs具有良好的分离能力。所有配合物在真空中的F值最小,说明受体在真空中对R/S-PFFs的选择分离能力最弱。受体-客体配合物在水中的F值最大,这表明使用水作为溶剂对R/S-PFFs具有较高的选择性。受体-客体配合物在甲苯中对R/S-PFF的选择性最低,但考虑到配体在甲苯中的结合能较大,甲苯依然是萃取溶剂的选择之一。S-PFF-Uranyl-PMMT的F值比R-PFF-Uranyl-PMMT的要大一些,表明了受体对S型客体的识别和分离能力比R型客体更强。
3 结 论
本文从理论上研究Uranyl-PMMT对R/S-丙溴磷的络合作用及对映选择性识别能力。基于密度泛函理论计算了受体、客体及受体-客体络合物的Mayer键级、AIM拓扑性质、定域化轨道指示函数(LOL)、前线分子轨道、红外光谱和吉布斯自由能变。研究结果表明,PMMT可与铀酰离子形成稳定的不对称受体,且对S型丙溴磷有更强的络合能力。热力学性质研究进一步表明,在标准状况(298.15 K、1.01×102kPa)下,受体与客体在水、乙醇、正辛醇、丙酮和甲苯中都能自发地发生络合反应。Uranyl-PMMT在以上4种有机溶剂中对映选择性系数均超过90%,尤其在极性的水、乙醇和丙酮中,Uranyl-PMMT对手性丙溴磷的对映选择性系数均在99%以上。
猜你喜欢
广州化工(2022年19期)2022-11-09 11:30:46
广州化工(2022年18期)2022-10-22 10:27:00
潍坊学院学报(2021年2期)2021-07-22 07:59:12
高等学校化学学报(2021年7期)2021-07-11 16:25:48
化学与生物工程(2021年2期)2021-02-25 12:56:46
无机材料学报(2020年2期)2020-03-08 14:05:54
硅酸盐通报(2020年1期)2020-02-25 10:01:30
分析化学(2019年3期)2019-03-30 10:59:24
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:34:54
核科学与工程(2015年2期)2015-09-26 11:57:21
