
两个有机锡杂环羧酸酯配合物的合成、结构及抗癌活性
2022-08-09 03:49周玉林张复兴朱小明徐赛男赵邠婕陈镜姣李芳芳
无机化学学报 2022年8期
周玉林 张复兴 朱小明 田 婕 徐赛男 赵邠婕 陈镜姣 李芳芳 邓 欣
(衡阳师范学院化学与材料科学学院,功能金属有机化合物湖南省重点实验室,金属有机新材料湖南省普通高等学校重点实验室,湘江上游重金属污染监测与治理湖南省工程研究中心,衡阳 421008)
有机锡配合物由于具有结构的多样性和应用的广泛性而备受人们广泛关注[1-6]。特别是有机锡配合物良好的抑制癌细胞增殖活性的发现,为开发广谱、高效的抗肿瘤药物开辟了新的方向[7-9]。研究表明,许多有机锡配合物比目前临床上广泛使用的抗癌药顺铂的抗癌活性还要高出许多[10-15]。杂环羧酸是一类具有丰富结构和较强生物活性的良好配体,能与有机锡形成具有特色结构和特殊性能的有机锡配合物,引起了人们的兴趣,近年有了一些相关的研究的报道[16~20]。为了更系统地研究该类配合物,我们合成了二(邻溴苄基)锡二(2-吡啶甲酸)酯(1)和三(2-甲基-2-苯基丙基)锡 3-吲哚丁酸酯 (2),通过元素分析、红外光谱、核磁共振(1H、13C和119Sn)、X射线衍射(XRD)、热重分析(TGA)进行了表征,用X射线单晶衍射测定了晶体结构,对其结构进行量子化学从头计算,探讨了配合物分子的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。此外,我们还研究了这2个配合物的热稳定性和体外抗癌活性。
1 实验部分
1.1 仪器与试剂
所有试剂均为市售分析纯。合成反应在恒温磁力加热搅拌器上完成。配合物的熔点用北京泰克X-4数字显微熔点仪测定。红外光谱用Shimadzu FTIR8700(KBr压片,400~4 000 cm-1)光谱仪测定。元素组成用PE-2400型元素分析仪测定。核磁共振谱用Avance Ⅲ HD 500 MHz全数字化超导核磁共振谱仪(瑞士Bruker公司,TMS为内标)测定。XRD用Shimadzu X射线衍射仪测定,测试条件:Cu靶Kα1,λ=0.154 06 nm,工作电流30 mA,电压40 kV,测试角度(2θ)范围 5°~80°,扫描速度 2(°)·min-1。晶体分子结构用BrukerSmart Apex Ⅱ CCD单晶衍射仪测定。利用TG209F3热分析仪,在空气氛中,加热速度为20 ℃·min-1,气体流速为20 mL·min-1,在50~800℃范围内对配合物进行热重测试。
1.2 配合物的合成
配合物1:在50 mL的耐压反应瓶中,加入无水甲醇35 mL、乙醚2 mL,在电磁搅拌器上搅拌1 min后加入0.046 g(2 mmol)切成小片的金属钠,搅拌至金属钠反应完全。加入0.246 g(2 mmol)2-吡啶甲酸,继续搅拌5 min,再加入0.615 g(1 mmol)二(邻溴苄基)二溴化锡,密封反应瓶,在110℃下恒温搅拌反应3 h。冷却后过滤,除去不溶性固体,滤液放置后析出白色固体,用二氯甲烷-乙醇重结晶得1的无色晶体0.538 g,产率76.53%。熔点:156~158℃。元素分析(C26H20Br2N2O4Sn)计算值(%):C,44.38;N,3.98;H,2.84。实测值(%):C,44.52;N,4.01;H,2.86。IR(KBr,cm-1):3 038,2 945,2 839ν(C—H),1 703νas(COO),1 308νs(COO),552ν(Sn—C),492ν(Sn—N),472ν(Sn—O)。1H NMR(CDCl3,500 MHz):δ8.19~8.11(m,4H),7.85(t,J=7.5 Hz,2H),7.29(t,J=6.5Hz,2H),7.11(d,J=9.0 Hz,2H),6.95~6.88(m,4H),6.86(t,J=7.5 Hz,2H),2.91(s,4H)。13C NMR(CDCl3,125 MHz):δ164.00,145.40,143.69,140.93,139.62,132.13,129.61,127.45,127.04,125.50,125.14,123.12,35.42。119Sn NMR(CDCl3,186 MHz):δ-366.31。
配合物2:在50 mL的耐压反应瓶中,加入无水甲醇 40 mL、1.052 g(1 mmol)双(三(2-甲基-2-苯基丙基)锡)氧化物、0.406 g(2 mmol)3-吲哚丁酸,密封反应瓶,在110℃下恒温搅拌反应4 h。冷却后旋转蒸发除去部分溶剂,放置后析出白色固体,用二氯甲烷-乙醇重结晶得2的无色晶体1.068 g,产率74.12%。熔点:102~104℃。元素分析(C42H51NO2Sn)的计算值(%):C,69.95;H,7.13;N,1.94。实测值(%):C,69.68;H,7.10;N,1.98。IR(KBr,cm-1):3 057,2 955,2 909,2 862ν(C—H),1 626νas(COO),1 368νs(COO),556ν(Sn—C),470ν(Sn—O)。1H NMR(CDCl3,500 MHz):δ8.05(s,1H),7.69~7.67(m,1H),7.41~7.39(m,1H),7.32~7.12(m,17H),7.03(d,J=1.5 Hz,1H),2.85(t,J=7.0 Hz,2H),2.38~2.35(m,2H),2.08~2.05(m,2H),1.24~1.15(m,24H)。13C NMR(CDCl3,125 MHz):δ178.26,150.88,136.33,128.28,127.56,125.77,125.28,121.82,121.30,119.07,119.00,116.21,111.01,37.67,37.15,35.65,32.71,26.21,24.78。119SnNMR(CDCl3,186MHz):δ-187.52。
1.3 晶体结构测定
分别选取大小为0.23 mm×0.21 mm×0.20 mm和0.22 mm×0.21 mm×0.20 mm 的 晶 体 ,在 Bruker SMART APEX Ⅱ CCD单晶衍射仪上,采用经石墨单色化的MoKα射线(λ=0.071 073 nm),于296(2)K,以φ-ω扫描方式收集数据。可观察衍射点数分别为4 345和7 322(I>2σ(I)),用于结构分析和精修。衍射强度数据经多重扫描吸收校正,晶体结构中大部分非氢原子由直接法解出,其余部分非氢原子在随后的差值傅里叶合成中陆续确定,对所有非氢原子坐标及其温度因子采用全矩阵最小二乘法精修。由理论加氢法给出氢原子在晶胞中的位置坐标,对氢原子和非氢原子分别采用各向同性和各向异性热参数精修,全部结构分析工作在WINGX上调用SHELX-97程序完成。配合物的主要晶体学数据列于表1。

表1 配合物1和2的晶体学数据Table 1 Crystallographic data of complexes 1 and 2
CCDC:2133058,1;2133057,2。
1.4 配合物的体外抗癌活性测定
人肝癌细胞(HUH7)、人肺癌细胞(A549)、人表皮癌细胞(A431)、人结肠癌细胞(HCT-116)和乳腺癌细胞(MDA-MB-231)取自美国组织培养库,用含10%牛胎血清的RPMI1640(GIBICO,Invitrogen)作培养基,在CO2体积分数5%的培养箱内于37℃下培养,用MTT法检测细胞增殖与生长抑制情况,调整实验细胞数量使其在570 nm获得1.3~2.2的吸光度,将配合物测试药液(0.1 nmol·L-1~10 μmol·L-1)设置6个浓度,处理细胞72 h,每个浓度至少3个平行和3次重复实验,应用GraphPad Prism5.0软件统计分析确定半抑制率IC50。
2 结果与讨论
2.1 配合物的谱学性质
在红外光谱图中,配合物1在492和472 cm-1处出现了吸收峰,分别为Sn—N键和Sn—O键的吸收峰;配合物2在470 cm-1处出现了吸收峰,为Sn—O键的吸收峰。配合物1和2的羧基的不对称伸缩振动νas(COO)和对称伸缩振动吸收峰νs(COO)分别在1 703、1 308 cm-1和1 626、1 368 cm-1出现了尖锐吸收峰,其 Δν(νas(COO)与νs(COO)之差)均大于200 cm-1,表明配合物1和2中羧基氧都是以单齿形式与锡配位。
在1H NMR谱中,配合物1和2分别在δ=8.19~6.86和7.69~7.03间呈现多重峰,为芳环上质子的峰,δ=8.05为配合物2的N—H的峰,配合物1亚甲基氢峰的δ在2.91;配合物2中3-吲哚丁羧基的3个亚甲基氢分别在δ为2.86~2.84、2.38~2.35、2.08~2.05,1.24~1.15出现的峰对应2-甲基-2-苯基丙基中甲基和亚甲基的氢。在13C NMR谱中,配合物1和2羰基碳的δ分别在164.00和178.26处,芳环上碳分别在δ为145.40~123.12、150.88~111.01处出现了吸收峰。配合物1的亚甲基碳的δ在35.42,配合物2的烷基碳的δ出现在37.67~24.78。在119Sn NMR谱中,配合物1和2分别在δ为-366.31和-187.52处出现峰。以上红外和核磁数据结果与X射线单晶衍射测定的晶体结构相吻合。
2.2 晶体结构分析
配合物的主要键长和键角列于表2,配合物的分子结构见图1、图2。对于配合物1,由图1和结构参数可知,其为单锡核分子,分子中2个2-吡啶甲酸配体分子均是以羧基氧单齿和吡啶环氮与锡原子螯合配位,中心锡原子与2个氧原子、2个氮原子和2个苄基亚甲基碳原子相连,形成六配位的八面体构型。其中2个氧原子O1、O3和2个氮原子N1、N2处于赤道平面的4个位置,2个亚甲基碳原子C13、C20则占据了赤道平面两侧的轴向位置。处于赤道位置的4个原子之间的夹角分别为O1—Sn1—N2 79.8(3)°、O3—Sn1—N271.2(3)°、O1—Sn1—N172.2(3)°、O3—Sn1—N1 139.5(3)°。其夹角之和为 360.7°,与360°有偏差,说明处于赤道位置的4个原子未能很好共平面。处于轴向位置的2个原子C13和C20与处于赤道位置的4个原子的键角在101.3°~81.6°之间,均与90°有偏差,且处于轴向位置的键角C13—Sn1—C20为158.8°,与180.0°线性角相差很大。由此可知,分子中锡原子为畸变程度很大的八面体构型。

表2 配合物1和2的主要键长(nm)和键角(°)Table 2 Selected bond distances(nm)and bond angles(°)of complexes 1 and 2
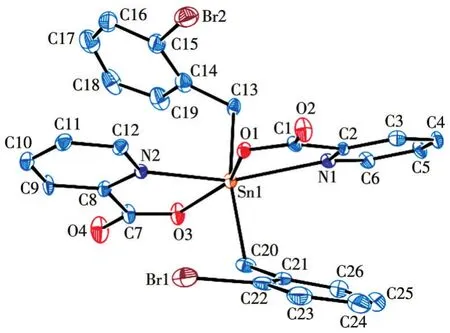
图1 配合物1的椭球概率15%的分子结构图Fig.1 Molecular structure of complex 1 with the ellipsoids drawn at the 15%probability level
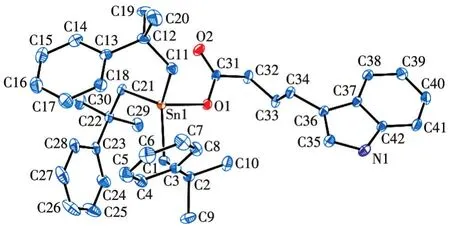
图2 配合物2的椭球概率15%的分子结构图Fig.2 Molecular structure of complex 2 with the ellipsoids drawn at the 15%probability level
对于配合物2,由图2和结构参数可知,其为单锡核分子,中心Sn原子与3个2-甲基-2-苯基丙基亚甲基C原子和1个羧基O原子相连形成四面体构型。由于配体3-吲哚丁酸基和2-甲基-2-苯基丙基的空间作用,致使配合物中3个Sn—C键的键长键角不等。配合物中Sn原子与2个羧基氧原子O1、O2之间的距离分别为0.207 3、0.300 7 nm,其中O1与Sn原子之间的距离小于Sn原子与O原子的共价半径之和(0.216 nm),而O2与Sn原子之间的距离远大于这2种原子的共价半径之和,说明配合物中只有一个羧基O原子与Sn很好地成键,而另一个羧基O原子未能与Sn原子成键。因此,配合物中羧基是以单齿形式与锡原子配位,生成四配位的变形四面体,与红外光谱测得的结果一致。
2.3 配合物的XRD分析
为了验证配合物的纯度,对配合物1和2进行了XRD测试。图3是计算机模拟的配合物1和2的单晶模拟XRD图和相对应的测定的XRD图。由图可知,两者大部分衍射峰的位置保持一致,从而表明合成的配合物1和2的单晶纯度较高。
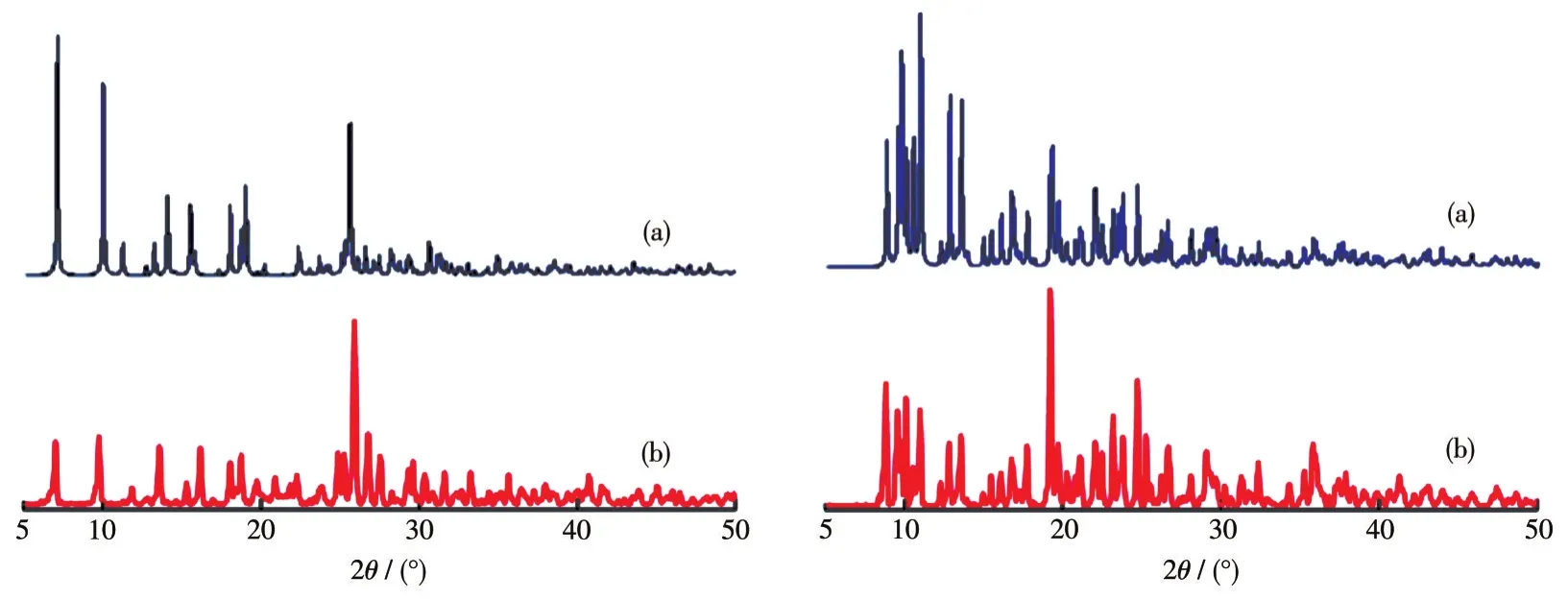
图3 配合物1和2的XRD图Fig.3 XRD patterns of complexes 1 and 2
2.4 量子化学研究
根据晶体结构的原子坐标,运用Gaussian 03W程序和B3lyp/lanl2dz基组水平,计算得到分子的总能量(ET)和分子轨道能量(EHOMO、ELUMO)。配合物1:ET=-1 442.351 327 8 a.u.,EHOMO=-0.229 70 a.u.,ELUMO=-0.083 85 a.u.,ΔELUMO-HOMO=0.145 85 a.u.。配合物 2:ET=-1 838.627 066 2 a.u.,EHOMO=-0.198 02 a.u,ELUMO=-0.006 80 a.u.,ΔELUMO-HOMO=0.191 22 a.u.。从体系能量和前沿轨道的能量来看,体系能量和前沿占有轨道的能量均较低,表明配合物分子结构具有一定的稳定性。但HOMO与LUMO的能量间隙ΔELUMO-HOMO均较小,说明配合物均较易失去电子而被氧化。
为探索配合物的电子结构与成键特征,将配合物分子轨道进行分析,用参与组合的各类原子轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。分别把配合物原子分为7部分,配合物1:(a)锡原子Sn;(b)配体羧基氧原子O;(c)配体羧基碳原子C1;(d)苄基亚甲基碳C2;(e)邻溴苯基碳原子和溴原子M1;(f)配体吡啶环碳原子和氮原子M2;(g)氢原子H。配合物2:(a)锡原子Sn;(b)配体羧基氧原子O;(c)配体羧基碳原子C1;(d)2-甲基-2-苯基丙基亚甲基碳C2;(e)苯基异丙基碳原子C3;(f)配体吲哚丙基碳原子和氮原子M;(g)氢原子H。前沿占有轨道和未占有轨道各取5个,计算结果如表3、表4和图4、图5所示。稳定性;二是2-甲基-2-苯基丙基亚甲基碳原子和羧基氧原子与锡原子具有良好的结合,分子中Sn—C键和Sn—O键较稳定。比较HOMO与LUMO的各类原子轨道成分,可以看出,当电子从HOMO激发到LUMO时,配体吲哚丁酰氧基上的电子通过锡原子向2-甲基-2-苯基丙基上转移,锡原子既是电子转移的桥梁也是电子转移的部分受体。
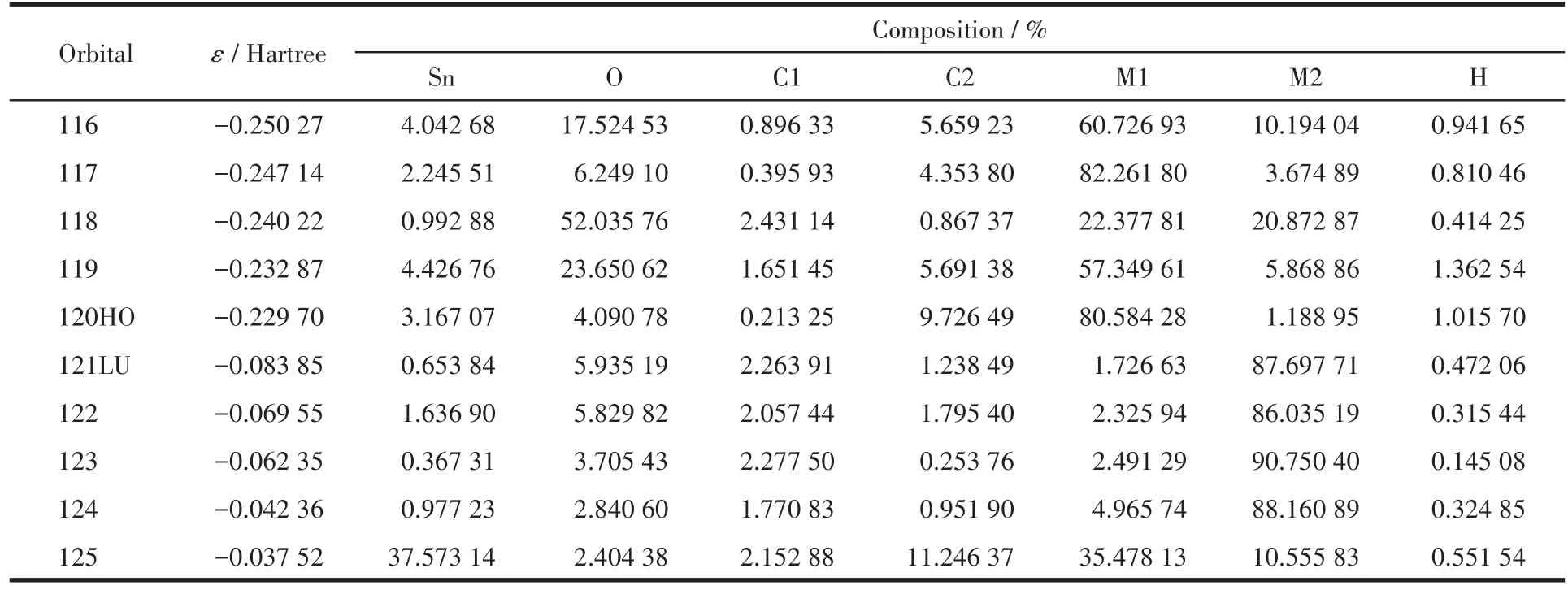
表3 配合物1的分子轨道组成Table 3 Calculated frontier molecular orbitals composition of complex 1
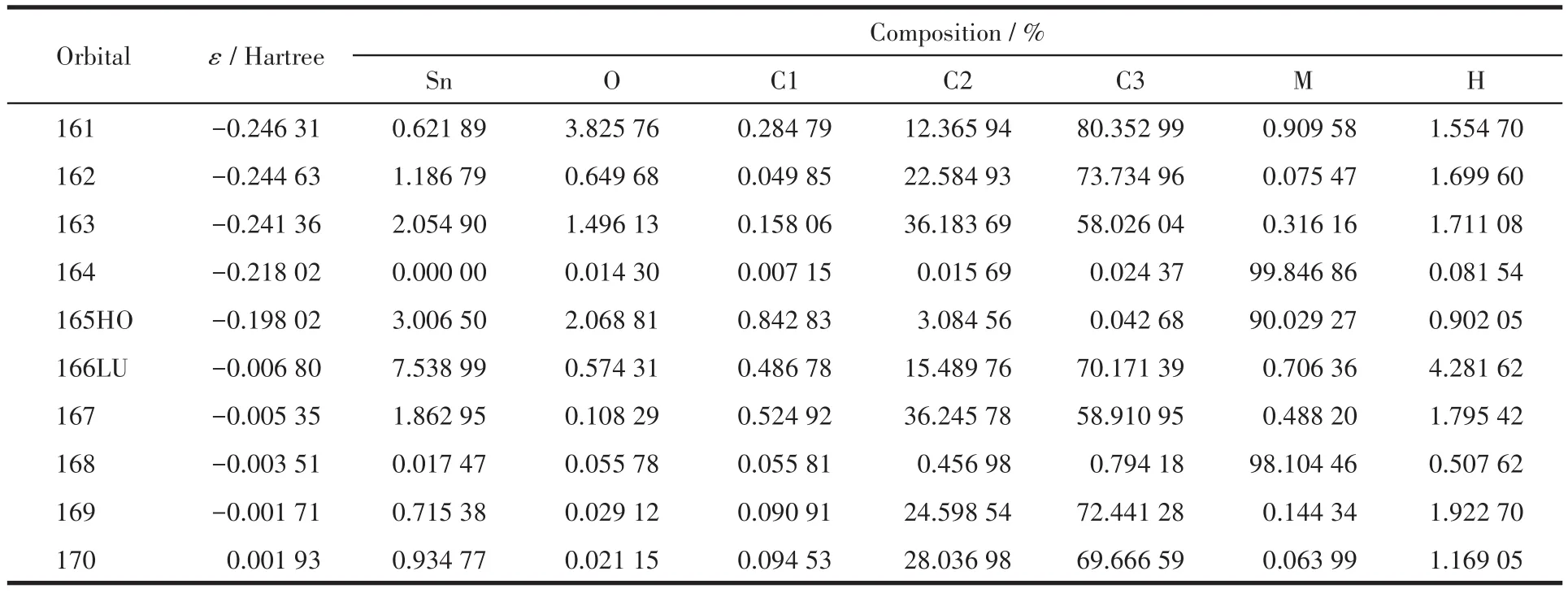
表4 配合物2的分子轨道组成Table 4 Calculated some frontier molecular orbitals composition of complex 2
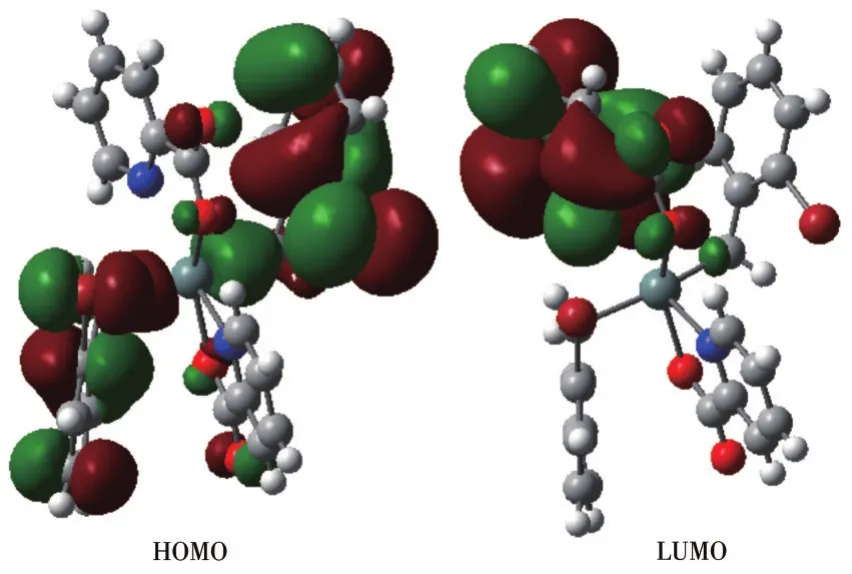
图4 配合物1的前沿分子轨道示意图Fig.4 Schematic diagram of frontier molecular orbital for complex 1
图5 配合物2的前沿分子轨道示意图Fig.5 Schematic diagram of frontier molecular orbital for complex 2
表3和图4显示配合物1的成键特征:前沿占有分子轨道中,对分子轨道贡献最大的是邻溴苯基碳原子,为80.58%;其次是邻溴苄基亚甲基碳原子、配体羧基氧原子和锡原子,分别为9.72%、4.09%和3.17%。说明:一是溴代苯环具有良好的共轭性和较强的稳定性;二是亚甲基碳原子和羧基氧原子与锡原子具有良好的结合,分子中Sn—C键和Sn—O键较稳定。比较HOMO与LUMO的各类原子轨道成分,可知,当电子从HOMO激发到LUMO时,邻溴苄基和锡原子上的电子向配体转移,配体的羧基既是电子转移的桥梁也是电子转移的部分受体,而配体的吡啶环则是电子转移的主要受体。
表4和图5显示配合物2的成键特征:前沿占有分子轨道中,对分子轨道贡献最大的是配体吲哚丙基碳原子和氮原子,为90.03%;其次是2-甲基-2-苯基丙基亚甲基碳、锡原子和羧基氧原子,分别为3.08%、3.01%和2.07%。说明:一是配体有较好的
2.5 热稳定性分析
配合物的热稳定性分析结果如图6所示。对于配合物1,在170℃之前几乎没有失重;从170℃起配合物开始缓慢失重,到220℃时开始快速失重,到370℃时失重变缓,至740℃时失重基本停止,残留质量最后稳定在20.79%。总计失重79.21%。残余物可认为是SnO2,与计算值21.44%基本吻合。对于配合物2,在280℃之前几乎没有失重;从280℃开始配合物缓慢失重,330℃时快速失重,到390℃时失重变缓,至655℃时失重基本停止,残留质量最后稳定在19.06%,总计失重80.94%。残余物可认为是SnO2,与计算值20.92%基本吻合。
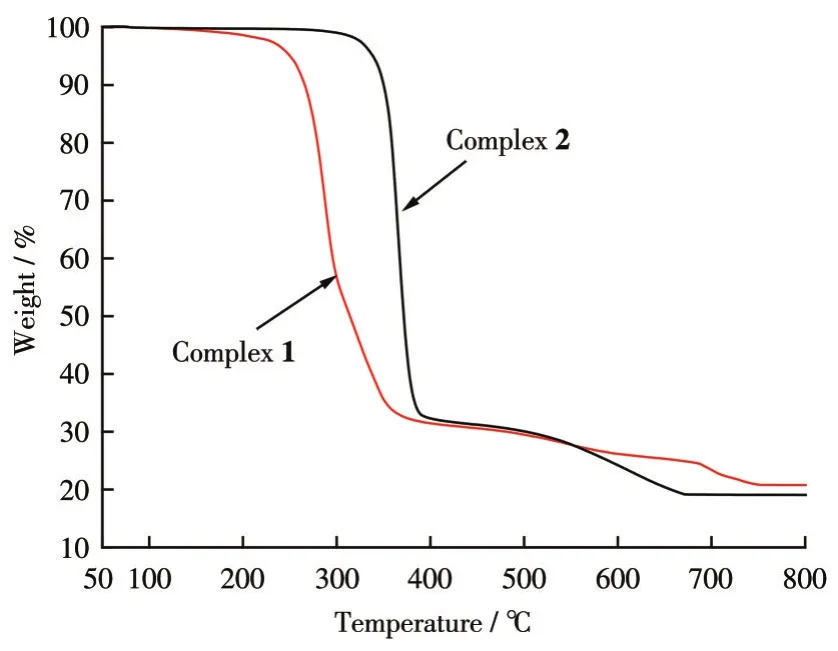
图6 配合物1和2的TGA曲线Fig.6 TGA curves of complexes 1 and 2
2.6 抗肿瘤活性
以顺铂对照,测试了配合物对癌细胞HUH7、A549、A431、HCT-116和MDA-MB-231的体外生长抑制活性,结果见表5。结果显示,配合物1和2对这5种癌细胞的抑制活性均比顺铂强。但由于2个配合物烃基和配体结构的不同,其抗癌活性也存在一些差异,除MDA-MB-231以外,配合物2的抑制活性均比1要强,它们更详细的生物活性有待进一步研究。
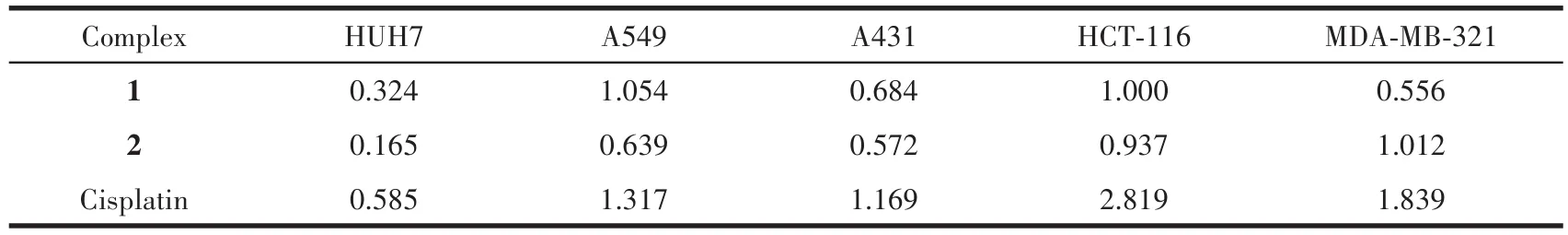
表5 配合物1和2对体外肿瘤细胞的IC50Table 5 IC50of complexes 1 and 2 on tumor cells in vitro μmol·L-1
3 结论
以甲醇为溶剂,用溶剂热法合成了2个有机锡杂环羧酸酯配合物:二(邻溴苄基)锡二(2-吡啶甲酸)酯(1)和三(2-甲基-2-苯基丙基)锡3-吲哚丁酸酯(2)。体外抗癌活性测试表明这2个配合物对人肝癌细胞(HUH7)、人肺癌细胞(A549)、人表皮癌细胞(A431)、人结肠癌细胞(HCT-116)和乳腺癌细胞(MDA-MB-231)的抑制活性均比顺铂强。
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
云南大学学报(自然科学版)(2022年3期)2022-05-25
宇航材料工艺(2020年1期)2020-03-26
中小学班主任(2019年12期)2019-09-10
新课程·中旬(2016年12期)2017-05-08
中学生数理化·高二版(2017年2期)2017-04-19
科技创新导报(2016年30期)2017-03-15
湖北农业科学(2014年9期)2014-08-08
润滑油(2009年4期)2009-01-18
