
阿哌沙班的中试合成工艺研究
2022-08-08 12:06王硕冰王恩慧崔闻宇吕江维李文兰王钝
当代化工研究 2022年14期
*王硕冰 王恩慧 崔闻宇 吕江维 李文兰* 王钝
(1.哈尔滨商业大学药学院 黑龙江 150076 2.沈阳药科大学制药工程学院 辽宁 110016)
阿哌沙班(apixaban,1),化学名为l-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代哌啶-1-基)苯基]-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酰胺,CAS:503612-47-3,其结构式如图1所示[1]:

图1 Apixaban结构式
目前已有多篇文献对1的合成进行报道[2-5],但这些合成路线大多都存在原料昂贵、反应时间长、后处理繁琐、操作不便、产品收率低等问题,不适于工业化生产。本研究在查阅相关文献和前期小试工艺研究的基础上,确定了如下中试合成工艺路线:以关键中间体5,6-二氢-3-(4-吗啉基)-1-[4-(2-氧代-1-哌啶基)苯基]-2(1H)-吡啶酮(A)和2-氯-2-[2-(4-甲氧基苯基)亚肼基]乙酸乙酯(B)为起始原料,经[1+3]偶极环加成反应生成1-(4-甲氧基苯基)-6-[4-(2-氧代哌啶-1-基)-苯基]-7-氧代-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酸乙酯(即阿哌沙班乙酯,C),C经酰胺化反应即可制得阿哌沙班粗品(记为1a),粗品1a经两次重结晶纯化得阿哌沙班成品(第一次重结晶所得粗品1记为1b,第二次重结晶所得成品记为1)。1的中试合成工艺路线图2所示:

图2 1的中试合成工艺路线
1.工艺研究
(1)阿哌沙班乙酯(C)的合成工艺研究
在C的制备中,滴加碱性催化剂三乙胺的时间以及加毕后的反应温度对C的收率和质量有较大影响。在进行多批次不同条件的中试后,发现当滴加三乙胺的时间控制在2~4h内、加毕后反应温度控制在87~93℃内时,C的收率(指重量收率,下文相同)以及纯度可以达到最高水平,分别为85.7%和99.92%。而当超出该最佳工艺参数范围时,会导致反应不完全或杂质增多。
(2)阿哌沙班粗品(1a)的合成工艺研究
在1a的制备过程中,除水是否充分是影响这一步骤产品收率和质量的关键因素。反应体系中所残留的水可以与后续加入的甲醇钠反应生成氢氧化钠,而氢氧化钠则可使得阿哌沙班乙酯(C)中酯基发生水解,生成主要有关物质阿哌沙班羧酸(化学名为4,5,6,7-4H-1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代-1-哌啶基)苯基]-1H-吡唑并[3,4-c]吡啶-3-羧酸,其结构如图3所示),它很难再转化为阿哌沙班,会残留在最终产品中成为主要杂质。经实验摸索,采用原甲酸三甲酯和三氟乙酸作为除水试剂,同时需要足够的除水时间和除水温度来保证反应体系中残留水分被充分除去。在进行多批次不同除水温度和除水时间的的中试后,确定最佳除水温度范围为45~50℃、最佳除水时间为40~45min。除此之外,反应时间对产品收率和质量同样有较大影响,反应时间过长会导致杂质增加,而时间过短则会使得原料转化不完全,产品产率降低。经多批次中试试验后确定35~45min为为最佳反应时间范围。当各关键工艺参数控制在最佳范围内时,1a收率可达90.4%,纯度则可达到99.77%,主要杂质阿哌沙班羧酸含量为0.13%。

图3 主要有关物质阿哌沙班羧酸的生成
(3)阿哌沙班粗品(1b)的合成工艺研究(第一次重结晶)
考虑到上步中可能存在除水不充分导致杂质阿哌沙班羧酸的生成,而且所使用的甲醇钠本身会含有一定量的氢氧化钠杂质,因此,为保证最终产品质量符合药典标准,对粗品进行重结晶是十分必要的。经实验摸索确定采用冰醋酸体系对阿哌沙班粗品(1a)进行重结晶。在本次重结晶过程中,养晶时间对重结晶结果有显著影响,养晶时间太短会导致产品收率显著降低,因此需要保证足够的养晶时间以达到最高产率。多批次中试试验表明当养晶时间达到6h时,1b收率最高,可达58.3%,纯度亦可达到99.92%,主要杂质阿哌沙班羧酸降低至0.06%;继续延长养晶时间对产品收率和质量无明显增益,因此确定最佳养晶时间为6h。
(4)阿哌沙班成品(1)合成工艺研究(第二次重结晶)
经实验摸索确定采用V(甲醇):V(水)=4:1为第二次重结晶的溶剂体系。在第二次重结晶过程中,溶解温度对本次重结晶收率有较大影响,若温度太低则会导致1b溶解不充分,从而使成品收率降低。在进行多批次中试试验后确定65~75℃为最佳溶解温度范围,在此范围内,重结晶收率为78.9%,1的纯度可达99.93%,主要杂质阿哌沙班羧酸继续降低至为0.04%。
2.阿哌沙班晶型研究
专利US2006069258 A1[6]报道了阿哌沙班具有两种晶型:非溶剂晶型N-1和二水合物晶型H2-2,并给出了各晶型相关参数。非溶剂晶型N-1为上市阿哌沙班的晶型。本文对以上制得的阿哌沙班进行了固体核磁及X-射线粉末衍射(X-ray powder diffraction,XRPD)分析,所得数据与专利文献中相关参数进行对比,确定通过本工艺路线所制备的阿哌沙班成品晶型为非溶剂晶型N-1。
固体核磁数据对比如表1所示:
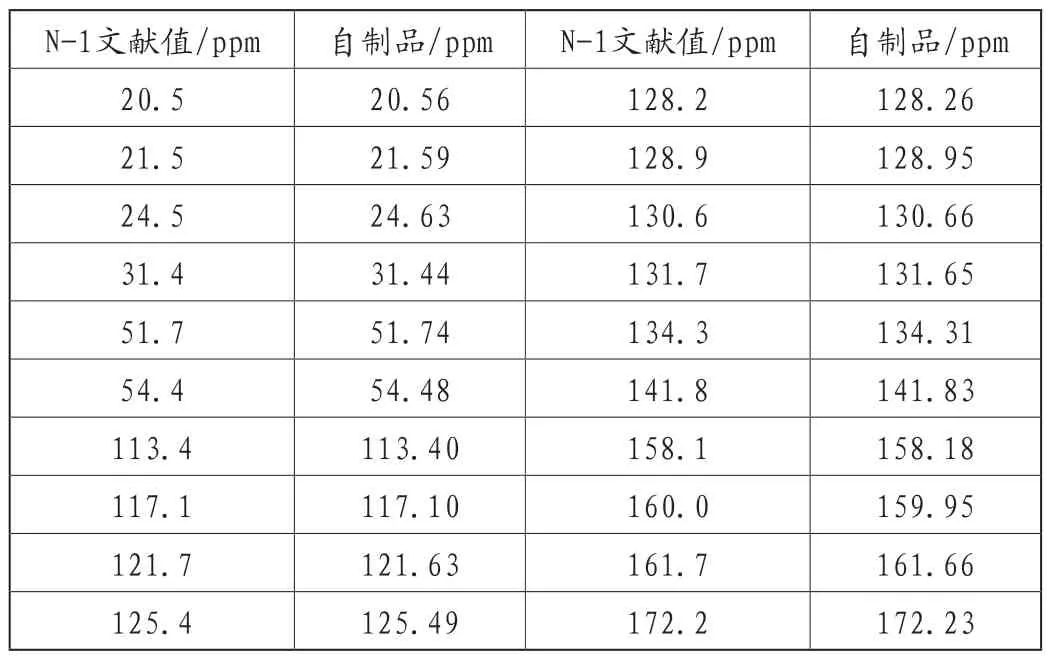
表1 阿哌沙班成品固体核磁数据对比
XRPD中主要2θ衍射角数据对比如表2所示:

表2 XRPD中阿哌沙班成品的X-射线粉末衍射主要2θ衍射角数据对比
3.阿哌沙班稳定性研究
本研究分别在25℃、30℃以及40℃下对以上制得的阿哌沙班成品进行了为期1~6个月的稳定性试验,在试验过程中除工艺杂质阿哌沙班羧酸外未见其它降解杂质产生,表明通过本工艺路线制得的阿哌沙班成品具有较好的稳定性,能够在药品期限内进行保存与运输。
4.实验部分
核磁共振1H-NMR采用Bruker ARX-400型核磁共振分析仪进行测定(TMS为内标)。X-射线粉末衍射采用日本理学Rigaku D/max-2400 X-射线粉末衍射仪进行测定,测试条件:CuKα1辐射,石墨单色器,40kV,150mA,8°/min,0.02°步长。玻璃反应釜购自南通普瑞科技有限公司,型号为50L FPGR-50,80L FPGR-80。
原料5,6-二氢-3-(4-吗啉基)-1-[4-(2-氧代-1-哌啶基)苯基]-2(1H)-吡啶酮(A)和2-氯-2-[2-(4-甲氧基苯基)亚肼基]乙酸乙酯(B)均购自安润医药科技有限公司。
(1)1-(4-甲氧基苯基)-6-[4-(2-氧代哌啶-1-基)-苯基]-7-氧代-4,5,6,7-四氢-1H-吡唑并[3,4-c]吡啶-3-甲酸乙酯(C)
开启搅拌(200~250r/min),向50L玻璃反应釜内加入2.5kg A,2.0kg B,21.78L甲苯,升温至90℃。向反应釜内滴加2.10L三乙胺,控温90℃,加毕计时搅拌(200~250r/min)反应3h。降温至室温,向反应釜内加入7.56L乙酸乙酯,降温至0~10℃,向反应釜内加入9.8L 4mol/L盐酸。加毕,0~20℃下计时搅拌(200~250r/min)4h。抽滤,滤饼用7.00L无水乙醇洗涤,抽滤后滤饼放入真空干燥箱干燥8h,得C(2.16kg,85.71%)。1H-NMR(400MHz,DMSO-d6)δ:1.422~1.446(t,J=7.2Hz,2H),1.962(m,4H),2.627(m,2H),3.323(m,2H),3.616(m,2H),3.808(s,3H),4.133(m,2H),4.446~4.480(q,J=6.4,2H),6.904~6.917(d,J=7.8,2H),7.267(s,2H),7.354(s,2H),7.468~7.481(d,J=7.8,2H)。
(2)阿哌沙班粗品(1a)
开启搅拌(200~250r/min),向50L玻璃反应釜内加入2.1kg C,8.36L甲酰胺、16.72L二甲基甲酰胺,升温至45℃,加入450mL原甲酸三甲酯、150mL三氟乙酸,加毕计时搅拌(200~250r/min)40min,向反应釜内加入2.7L甲醇钠/甲醇溶液,加毕计时搅拌(200~250r/min)40min,向反应釜内加入850mL纯化水,加毕,降温至0~10℃,计时搅拌(200~250r/min)4h;维持温度0~10℃,向反应釜内加入纯化水15.8L,加毕计时搅拌(200~250r/min)1h。抽滤,滤饼用乙醇洗涤两次,每次用量为5.00L,所得滤饼放入真空干燥箱干燥10h,得1a(1.89kg,90.43%)。
(3)阿哌沙班粗品(1b)
向50L玻璃反应釜内加入1.8kg 1a,18.0L冰醋酸,8.0L纯化水,室温搅拌(150~200r/min)至澄清。向反应釜内加入90g活性炭,加毕计时搅拌(150~200r/min)30min。过滤,转入50L玻璃反应釜中;向该反应釜内加入8.00L纯化水,室温下搅拌(150~200r/min)养晶8h。抽滤,滤饼用无水乙醇洗涤两次,每次用量为5.00L。所得滤饼放入真空干燥箱干燥10h,得1b(1.05kg,58.3%)。
(4)阿哌沙班成品(1)
向80L玻璃反应釜内加入0.9kg 1b、32.4L无水甲醇,8.0L纯化水,升温至65~75℃,搅拌(100r/min)溶解。趁热过滤,转入另一80L反应釜中,降温至室温,搅拌养晶5h。抽滤,滤饼用无水乙醇洗涤两次,每次用量5.0L,所得滤饼放入真空干燥机干燥10h,得1(0.71kg,78.9%)。1H-NMR(400MHz,DMSO-d6)δ:1.867(m,4H),2.410(t,2H),3.222(t,2H),3.609(t,2H),3.819(s,3H),4.067(t,2H),7.016(d,2H),7.292(d,2H),7.366(d,2H),7.467(s,1H),7.521(d,2H),7.737(s,1H)。
5.结果与讨论
本研究在前期小试工艺研究的基础上进行了阿哌沙班的中试合成工艺研究,对各步骤中所涉及到的影响因素(如反应条件、设备规格、后处理方法等)进行了探索,确定了各步骤中对产品质量和收率有显著影响的关键工艺参数,并对其进行优化,找到最佳工艺参数范围,从而实现更高的收率和更好的产品质量。
成品中的主要有关物质为阿哌沙班羧酸,是在酰胺化反应步骤中由阿哌沙班乙酯水解产生,通过减少反应体系中的水份可减少该杂质的生成;本工艺在反应体系中适量的原甲酸三甲酯和三氟乙酸作为除水试剂,有效控制了该杂质的生成。阿哌沙班粗品该杂质含量仅为0.13%,通过两次重结晶后降低为0.04%,在稳定性试验中成品放置6个月后该杂质的含量未见增加。
经本文优化后的中试合成工艺路线所用原料与试剂方便易得、反应条件温和,产品总收率为35.7%,纯度可达99.93%,质量稳定,表明该路线适合用于工业化生产。
猜你喜欢
广州化工(2022年20期)2022-12-01
再生资源与循环经济(2022年9期)2022-11-20
食品研究与开发(2022年9期)2022-05-17
中国氯碱(2022年2期)2022-03-21
杭州师范大学学报(自然科学版)(2021年6期)2021-12-07
化学与粘合(2021年4期)2021-08-02
铜业工程(2021年1期)2021-04-23
天津化工(2021年3期)2021-01-08
宇航材料工艺(2020年1期)2020-03-26
农药科学与管理(2019年8期)2019-11-23
