
壳聚糖包裹CdSe量子点组装体的水相可见光催化CO2还原
2022-08-06 04:39:32任颖异
高等学校化学学报 2022年7期
夏 雾,任颖异,刘 京,王 锋
(1.华中科技大学化学与化工学院,能量转换与储存材料化学教育部重点实验室,材料化学与服役失效湖北省重点实验室,武汉 430074;2.吉首大学化学化工学院,吉首 416000)
光催化二氧化碳(CO2)还原是人工光合作用研究的重要组成部分. 人工光合作用体系利用太阳能驱动CO2转化为高附加值的太阳燃料(如一氧化碳、甲烷、甲醇等),可实现CO2的资源化利用[1~3]. 发展高效、高选择性的CO2光催化剂/体系是当前该领域研究的主要任务[4~8]. 半导体纳米材料因其独特的光物理和光化学性质而被应用于构筑光催化CO2还原体系[9~11],其中,Ⅱ-Ⅵ族硒化镉量子点(CdSe QDs)作为光催化剂可将CO2催化还原为CO[10,12~14]. Frei 等[13]利用瞬态衰减全反射-傅里叶变换红外光谱(ATR FTIR)对CO2在CdSe QDs表面的光催化还原过程进行了研究,发现CdSe QDs表面吸附两分子CO2后形成Cd—C(=O)OCO2−中间体[吸收峰出现在(1575±5)cm−1处]的能量比CO•2−低84 kJ/mol,在热力学上更容易被还原,该中间体接收CdSe量子点导带电子后进一步被还原为CO. Wu等[12]对具有CO2催化活性的CdSe QDs(111)晶面进行了DFT计算,结果表明,与Cd—O和Cd—C的相互作用相比,CO2倾向于通过氧原子嵌入QDs 表面Se 空位中被活化,再形成Cd-COOH*中间体,最后分解产生CO,且CdSe/CdS QDs壳层中S空位可以起到相同的作用. CdSe/CdS QDs在三乙胺(TEA)/N,N-二甲基甲酰胺(DMF)溶液中光催化CO2为CO 的效率达到412.8 mmol·g−1·h−1,选择性大于96%. Yanagida 等[15]发现,向CdS QDs的DMF溶液中加入略微过量的Cd2+能够提高体系的光催化活性,而过量H2S的加入抑制了CO的产生,据此推测表面Cd2+为光催化活性位点,过量Cd2+加入会造成S空位的产生,并利用原位CdK-edge X 射线吸收精细结构谱(EXAFS)证实了S空位的存在. 在光照下,CdS QDs导带电子将吸附在其表面的CO2还原为,其中CO2的C原子与Cd2+结合,其中一个氧原子进入CdS表面的S空位中进一步结合第二个CO2分子后形成Cd—C(=O)OCO−2中间体,并最终还原产生CO和上述研究表明,CdSe或CdS量子点表面的Cd2+是活化CO2分子的活性位点. 基于此,本课题组[14]通过调节CdCl2和Na2SeSO3的投料比,并在酸性条件下剥离QDs表面3-巯基丙酸(MPA)配体得到去配体表面富镉(LRCR)-CdSe QDs,以达到丰富并暴露活性位点的目的. FTIR、X 射线光电子能谱(XPS)和表面Zeta电势测试结果显示,随着n(CdCl2)∶n(Na2SeSO3)由2∶1 提高至8∶1,LRCR-CdSe QDs 表面Cd/Se 摩尔比由1.57 提高至6.53,QDs 表面呈富镉状态. 在TEA/DMF 溶液中,LRCR-CdSe QDs 光催化CO2还原的效率随QDs 表面Cd/Se比例增加而不断提高,最高光催化速率达到789 mmol·g−1·h−1(产物CO的选择性为95%).
上述基于CdSe QDs催化剂的CO2还原体系表现出优异的光催化性能,但均在有机溶剂中进行. 使用非质子性有机溶剂既可有效降低溶剂中质子的浓度;还能提高溶剂中CO2的溶解度(25 ℃时,CO2在DMF中的溶解度约为H2O中的6倍[16]),从而提高光催化CO2还原效率及产物的选择性. CO2在水中的溶解度低且质子还原反应的竞争性强,因此以CdSe QDs和CdS QDs光催化剂进行水相光催化CO2还原的体系较少且催化性能并不理想[12~14,17~19]. 水是优良绿色溶剂,且光合作用的最终目的是以水为电子供体形成全反应体系,因此发展高效、高选择性的水相二氧化碳光催化还原体系具有重要意义.
壳聚糖(chitosan)是甲壳素(chitin)脱乙酰化后的多糖类天然高分子,可以看作是纤维素C2位上羟基被氨基取代的产物,在自然界中的储量丰富[20,21],具有良好的生物相容性[22~24]. 壳聚糖的高分子骨架具有一定的疏水性,其结构单元中的氨基可以和量子点表面金属配位,从而将量子点引入其高分子组织结构中,同时,氨基可以可逆地与CO2反应生成氨基甲酸盐,从而增强CO2的吸附[22,25~29]. 基于上述壳聚糖的结构和性质特点,我们希望通过壳聚糖中氨基与MPA-CdSe QDs 表面Cd2+的配位作用形成MPA-CdSe@chitosan组装体,利用壳聚糖中丰富的氨基和疏水性结构为量子点提供富CO2、疏水的微环境,从而通过调控CdSe量子点的表/界面环境来提升CdSe QDs在水溶液中光催化CO2还原的效率和产物的选择性[30~33].
1 实验部分
1.1 试剂与仪器
亚硫酸钠(纯度99.0%)、氢氧化钠(纯度99.0%)、盐酸(质量分数36.0%)和抗坏血酸钠(L-Ascorbic Acid Sodium,NaHA,纯度99.0%),国药集团化学试剂有限公司;硒粉(Se,纯度99.9%),上海麦克林生化科技有限公司;氯化镉半五水合物(CdCl2∙2.5H2O,纯度99.9%)和3-巯基丙酸(3-Mercaptopropionic acid,MPA,纯度98.0%),上海阿拉丁生化科技股份有限公司;壳聚糖(脱乙酰度>95.0%)、氨基葡糖糖盐酸盐(纯度98.0%)和纤维素,上海迈瑞尔化学技术有限公司;氢气(纯度99.99%)、一氧化碳(纯度99.9%)、氩气(纯度99.999%)、二氧化碳(纯度99.99%)和甲烷(纯度99.99%),武汉中鑫瑞远气体有限公司.
RE-52AA型旋转蒸发仪,上海亚荣生化仪器厂;H1850型高速离心机,湖南湘仪离心机有限公司;Fiveeasy plus型pH计,Mettler-Toledo国际股份有限公司;UV-2600型UV-Vis光度计、RF-6000型荧光分光光度计和UV-3600型紫外-可见漫反射光谱仪,日本岛津公司;ESCALAB 250Xi型X射线光电子能谱仪(XPS),上海赛默飞世尔科技有限公司;Empyrean 型X 射线衍射仪(XRD),荷兰帕纳科公司;Talos F200X 型场发射透射电子显微镜(TEM),荷兰FEI 公司;Invenio-R 型傅里叶变换红外光谱仪(FTIR),德国Bruker公司;IC 861型离子色谱,瑞士万通有限公司;3 W LED光源,定制;GC7900型气相色谱仪,上海天美科学仪器有限公司;SGH-300A型氢气发生器和WJK-2LB型空气泵,杭州捷岛科学仪器有限公司.
1.2 实验过程
1.2.1 MPA-CdSe QDs 的合成 参照文献[34~36]方法合成MPA-CdSe QDs. 将189.0 mg(1.5 mmol)Na2SO3溶解于100.0 mL 去离子水中,再加入40.0 mg(0.5 mmol)硒粉,在N2气氛围下于130 ℃加热回流. 反应结束后自然冷却至室温,得到无色透明的Na2SeSO3溶液. 将其转移至棕色试剂瓶中,于N2气氛围避光保存备用.
将46.0 mg(0.25 mmol)CdCl2∙2.5H2O溶解于190.0 mL去离子水中,再加入上述10.0 mL NaSeSO3溶液和26.0 μL(0.30 mmol)MPA,使用NaOH溶液(1.0 mol/L)和HCl溶液(1.0 mol/L)调节溶液pH值至9.0~11.0. N2气氛围下于130 ℃加热回流,溶液由无色透明逐渐转变为橙黄色. 反应结束后,将反应溶液浓缩至40.0 mL,加入异丙醇(IPA)沉淀、离心,重复3次后收集沉淀,真空干燥得到MPA-CdSe QDs蛋黄色固体粉末.
1.2.2 MPA-CdSe@chitosan组装体光催化CO2还原 以MPA-CdSe@chitosan组装体在室温条件下光催化CO2还原为例:向硬质玻璃管中分别加入0.5 mg(0.1 mg/mL)MPA-CdSe QDs、19.8 mg(20.0 mmol/L)NaHA 和5.0 mg(1.0 mg/mL)壳聚糖,溶剂为H2O,溶液的总体积为5.0 mL,至少制备两组平行样品.向上述样品中持续鼓入高纯度的CO2气体25 min,再加入500 μL高纯度CH4作为内标物. LED光源的最大波长为450 nm(λmax=450 nm). 使用配备火焰离子化检测器(FID)和热导检测器(TCD)的GC7900型气相色谱仪分别对CO,H2及CH4等气体产物或内标物进行定量分析. TDX-01型填充柱[80/100目硅氧烷填料,2 m(柱长)×3 mm(外径)×2 mm(内径)]为分析柱,Ar为载气. 柱箱温度为110 ℃,进样口温度为120 ℃,TCD设定温度为150 ℃,FID设定温度为360 ℃.
1.2.3 CO2溶解量测试 25 ℃恒温条件下,向25 mL密封试管中加入5.0 mL去离子水后鼓入高纯度N2气25 min,再加入5.0 mL CO2和500 μL CH4(内标物). 利用气相色谱测试不同时间试管内气体中CO2的量,将CO2的初始量减去剩余量得到H2O中CO2的溶解量. 向去离子水中加入5.0和10.0 mg的壳聚糖进行测试作为对比.
2 结果与讨论
2.1 MPA-CdSe@chitosan组装体的构筑及表征
向含有0.5 mg(0.1 mg/mL)MPA-CdSe QDs 的水溶液中直接加入5.0 mg(1.0 mg/mL)壳聚糖(溶液体积为5.0 mL),量子点溶液由澄清变为浑浊,静置10 h后,溶液中的量子点随壳聚糖沉淀至比色皿底部(图1),出现絮凝现象[37],且上层溶液接近无色. 壳聚糖的加入使量子点从水相中分离,说明量子点与壳聚糖间存在相互作用. 利用TEM 可以直观地观察量子点的分布情况. 对比分散均匀的类球体形貌MPA-CdSe QDs[图2(A)],壳聚糖的加入使MPACdSe QDs沿聚合物链分布,沉积至壳聚糖上形成MPA-CdSe@chitosan组装体[图2(B)].

Fig.1 Comparison of MPA⁃CdSe QDs aqueous solution before and after adding chitosan

Fig.2 TEM images of MPA⁃CdSe QDs(A)and MPA⁃CdSe@chitosan assemblies(B)
分别对壳聚糖和不同光照时间的MPA-CdSe@chitosan组装体进行XRD测试. 由图3(A)可见,壳聚糖仅在19.9°处出现衍射峰,结晶性较差[38,39];MPA-CdSe 和MPA-CdSe@chitosan 在25.16°,42.07°和49.64°处的衍射峰分别与闪锌矿结构CdSe QDs 的(111),(220)和(311)晶面对应(JCPDS No.19-0191)[35]. 不同光照时间的MPA-CdSe@chitosan 组装体中均包含壳聚糖和量子点的特征衍射峰,且未发生变化,说明组装体稳定. 为了阐明MPA-CdSe@chitosan 组装体的形成机制,对上述样品进行FTIR 分析.图3(B)中对应于—NH2与金属离子相互作用的酰胺II 带(—N—H 的弯曲振动)的特征吸收峰(1527 cm−1)随光照时间的延长逐渐增强[40~46],说明MPA-CdSe@chitosan 组装体是通过壳聚糖中氨基与MPACdSe QDs量子点表面Cd2+配位而形成,且量子点与壳聚糖间的相互作用随时间延长而逐渐增强.

Fig.3 XRD patterns(A)and FTIR spectra(B)of MPA⁃CdSe(a),chitosan(b)and MPA⁃CdSe@chitosan assemblies after different irradiated time(c—e)
为进一步说明MPA-CdSe@chitosan组装体中两组分的相互作用,分别对MPA-CdSe QDs、壳聚糖和MPA-CdSe@chitosan 组装体进行XPS 分析,结果如图4 所示. MPA-CdSe QDs 表面Cd 的结合能为412.01 eV(Cd3d32)和405.30 eV(Cd3d52),内部Cd 的结合能为411.43 eV(Cd3d32)和404.69 eV(Cd3d52)[图4(A)][40,42,47,48]. MPA-CdSe@chitosan 组装体中表面Cd 的结合能为412.01 eV(Cd3d32)和405.28 eV(Cd3d52),内部Cd 的结合能为411.28 eV(Cd3d32)和404.55 eV(Cd3d52)[图4(C)],且新增一组Cd3d峰(Cd3d32,411.68 eV 和Cd3d52,404.95 eV)[40,42,47,48]. 相对于壳聚糖的N1s精细谱[图4(B),N1s,399.43 eV],MPA-CdSe@chitosan 组装体中新增结合能为400.40 eV 的N1s峰[40,42,47,48]. 上述结果与文献报道的Cd—N 键结合能一致,壳聚糖中氨基N 原子作为Lewis 碱增加Cd 的电子云密度,使MPA-CdSe QDs 的Cd3d结合能负移.
综合上述分析结果可知,壳聚糖中氨基与MPA-CdSe QDs 表面Cd2+配位生成Cd—N 键是形成MPA-CdSe@chitosan 组装体的关键,且MPA-CdSe@chitosan 组装体不溶于水. 根据MPA-CdSe QDs 和壳聚糖的性质,在MPA-CdSe@chitosan组装体中,壳聚糖将MPA-CdSe QDs包覆于其疏水结构中,使量子点处于疏水环境中,这十分有利于抑制质子还原为氢气,从而提高水相光催化CO2还原反应产物的选择性.

Fig.4 XPS of Cd3d of MPA⁃CdSe QDs(A),N1s of chitosan(B),Cd3d and N1s of MPA⁃CdSe@chitosan assemblies(C)
2.2 MPA-CdSe@chitosan组装体水相光催化CO2还原
2.2.1 MPA-CdSe@chitosan 组装体光催化CO2还原条件优化 质子还原产氢是水相光催化CO2还原的主要竞争反应,溶液pH 值影响MPA-CdSe@chitosan 组装体水相光催化CO2还原产物CO 的产生效率和选择性[49]. 由图5(A)可见,随着溶液pH值的增大,MPA-CdSe@chitosan组装体的光催化性能呈现先升高(pH:4.0~8.0)后降低(pH:8.0~12.0)的变化趋势. 根据Wu等[50]的研究结果,壳聚糖在酸性条件下易质子化,起到富集质子的作用,有利于氢气的产生. 而pH值由8.0提高至12.0时,CO的产生效率降低至7.2 mmol/g,对应的选择性为28.1%. 当pH=8时,MPA-CdSe@chitosan组装体光催化CO2还原产物CO 的产生效率达到33.8 mmol/g,对应的选择性为39.4%,分别是相同光催化条件下MPA-CdSe QDs[图5(B)]的169 倍和8.6 倍,表明壳聚糖的加入对MPA-CdSe QDs 光催化CO2还原性能产生了积极影响.

Fig.5 Effect of pH on photocatalytic CO2 reduction
壳聚糖通过氨基与量子点表面Cd2+配位,将量子点包裹于其疏水结构并提供疏水环境,从而影响还原产物CO的选择性. MPA-CdSe QDs和壳聚糖的相对含量对MPA-CdSe@chitosan组装体的光催化性能有明显影响,结果如图6(A)所示. MPA-CdSe@chitosan 组装体的光催化效率在壳聚糖的浓度为1.0 mg/mL时达到最大值,且还原产物CO的选择性随壳聚糖浓度的增加而降低. 壳聚糖的DRS图谱显示,其在小于500 nm 波长范围内有一定的吸收[图6(B)],与光催化使用的LED 光源波长范围(λmax=450 nm)有重叠,因此壳聚糖浓度的增加会产生滤光效应,导致产物CO的产生效率和选择性随壳聚糖浓度发生变化.
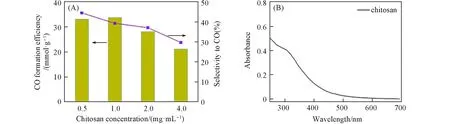
Fig.6 Effect of chitosan concentration on efficiency and selectivity of photocatalytic CO2 reduction(A)and DRS of chitosan(B)
2.2.2 MPA-CdSe@chitosan组装体的光催化稳定性 在最优光催化条件下进行长时间光照实验以考察MPA-CdSe@chitosan组装体的稳定性,结果如图7所示. 光催化60 h后CO产生效率为73.6 mmol/g,产物的选择性51.0%,且光催化速率在15 h时达到最大值3.5 mmol·g−1·h−1.
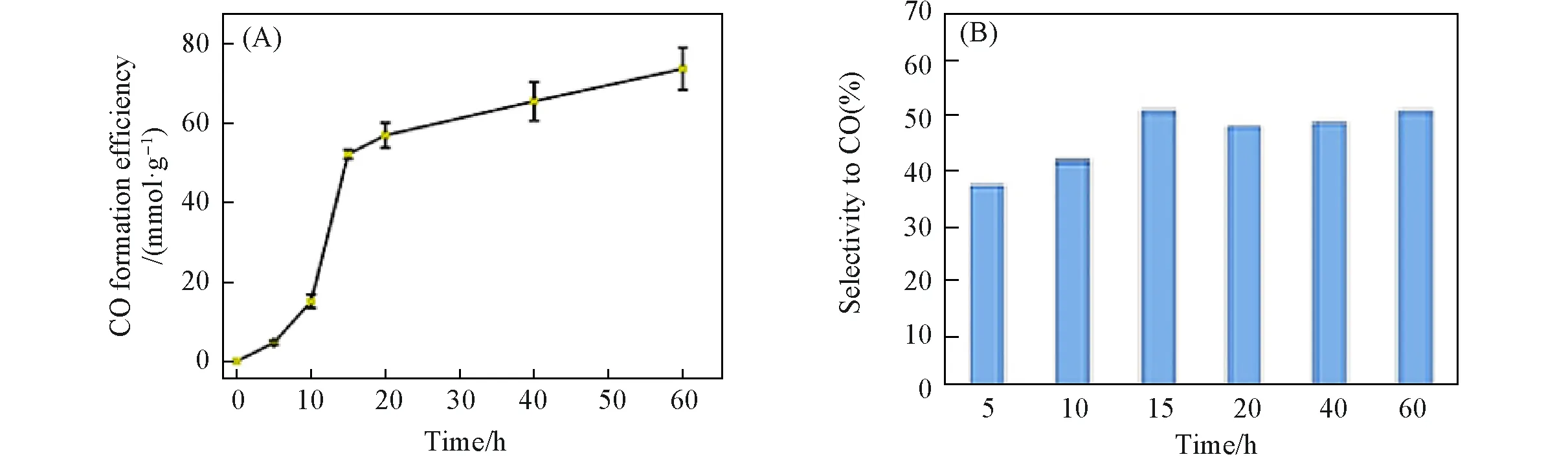
Fig.7 Duration experiment of photocatalytic CO2 reduction by MPA⁃CdSe@chitosan assemblies
2.2.3 MPA-CdSe@chitosan组装体光催化失活分析 实验结果表明,MPA⁃CdSe@chitosan组装体在光催化60 h后CO的产生效率不再提高,说明光催化体系中还原产物CO的量不再增加,体系无法继续进行光催化CO2还原,体系失活. 体系失活后溶液的pH值由8.0降至5.4. NaHA在光催化体系中作为电子牺牲体消耗MPA-CdSe QDs 价带中的空穴,生成脱氢抗坏血酸(DHA)并释放质子,使溶液的pH 值降低. 因此,在光催化CO2还原过程中,NaHA被消耗的同时会改变光催化反应溶液的酸碱环境. 光催化后MPA-CdSe@chitosan 组装体中Cd3d32和Cd3d52的结合能分别降低至411.64 eV 和404.89 eV,与之对应的表面Cd的结合能为411.98 eV(Cd3d32)和405.25 eV(Cd3d52),内部Cd的结合能为411.33 eV(Cd3d32)和404.59 eV(Cd3d52). 壳聚糖中的氨基因溶液pH 值降低而发生质子化(壳聚糖的pKa为6.3),对应结合能为401.60 eV 的N1s峰(图8)[40,42,47,48,51,52]. 图9给出不同光照时间的MPA-CdSe@chitosan 组装体样品的TEM 照片. 对比光催化前相对均匀分散的样品[图2(B)],量子点在光催化过程中趋向于沿壳聚糖聚合物链分布[图9(A)],两者间的相互作用不断增强. 光照60 h 后量子点的粒径明显变大,聚集较严重,但仍然保持了CdSe QDs 的核心结构[0.215 nm的晶格间距与d220吻合,图9(B)][35].
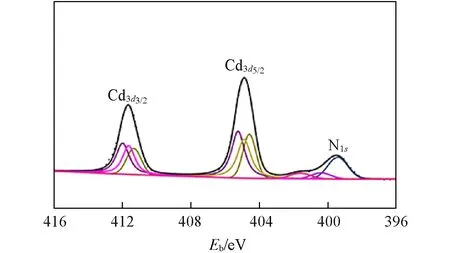
Fig.8 XPS of Cd3d and N1s in MPA⁃CdSe@chitosan assemblies after photocatalysis

Fig.9 TEM images of MPA⁃CdSe@chitosan assemblies after 5 h(A)and 60 h irradiation(B)
分析不同光照时间样品的Tauc 曲线可以获得光照前后量子点的带隙信息(图10)[53]. 随着光照时间的延长,量子点的带隙宽度逐渐减小,由光照前的2.53 eV逐渐减小至2.31 eV. 受量子限域效应的影响,量子点的带隙随粒径减小而逐渐增大,所以,可以进一步确认量子点在光催化过程中粒径有逐渐变大的趋势,结果与图9吻合. 量子点粒径变大会造成其比表面积减小,表面活性位点减少.
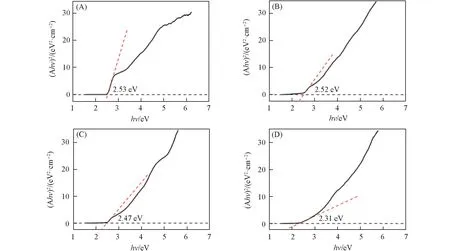
Fig.10 Tauc curves of MPA⁃CdSe QDs with different irradiated time
根据上述分析,随着光催化反应的进行,电子牺牲体NaHA会不断消耗,改变光催化溶液的酸碱环境. 在这个过程中,量子点的带隙不断减小,量子点的粒径出现不断增大的趋势,这些可能造成反应体系的失活.
2.3 MPA⁃CdSe@chitosan组装体光催化CO2还原机理
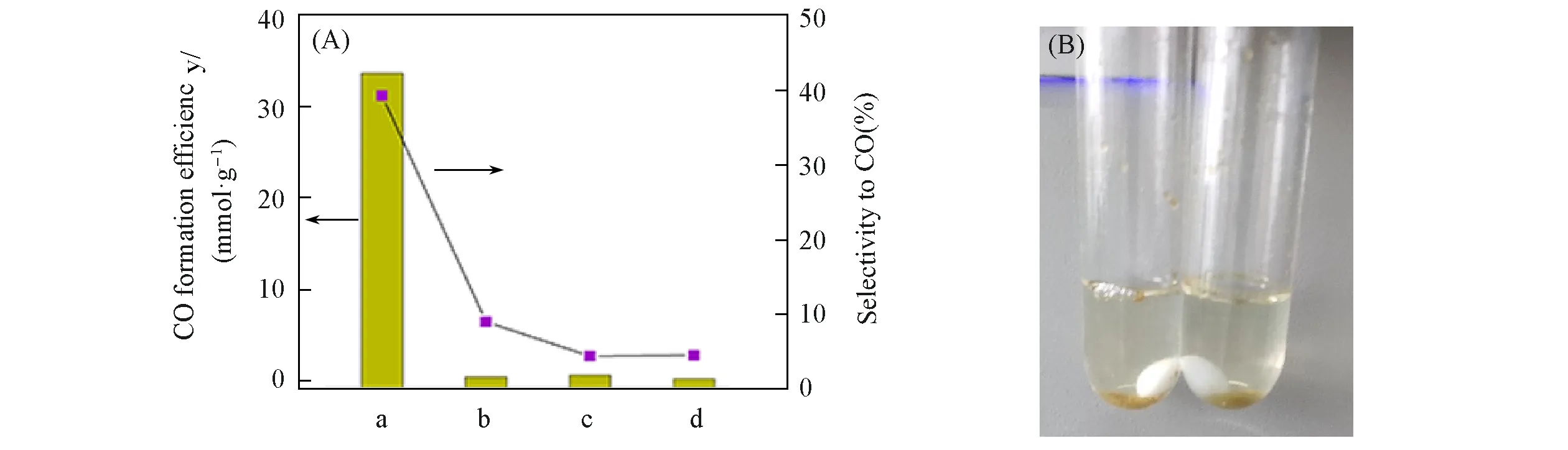
Fig.11 Effect of different additives of MPA⁃CdSe QDs on photocatalytic CO2 reduction(A)and photograph of MPA⁃CdSe@chitosan assemblies after 13 h irradiation(B)
壳聚糖与纤维素化学结构的差异主要为两者C2位上的基团不同,壳聚糖因糖苷键骨架和分子内及分子间氢键的存在,表现出一定的疏水性,在pH≥7 的溶液中基本不溶解[28,29]. 为进一步分析MPA-CdSe@chitosan组装体提升水相光催化性能(催化效率和选择性)的影响因素,选择D-氨基葡萄糖和纤维素进行对比实验[图11(A)]. 相对于单独使用MPA-CdSe QDs,MPA-CdSe@chitosan组装体的CO产生效率和选择性分别提高169倍和8.6倍. 光催化13 h后,上层清液为无色,MPA-CdSe@chitosan组装体以黄色沉淀存在于光反应试管底部[图11(B)],整体表现出疏水性. FTIR和XPS数据已经证实了壳聚糖中氨基与量子点表面镉离子的配位作用,所以选择水溶性的D-氨基葡萄糖(壳聚糖的结构单元)加入至MPA-CdSe QDs水溶液中进行光催化CO2还原,但产物CO的选择性仅为4.5%,说明仅有氨基与量子点表面Cd2+的作用并不能促进光催化性能提升. 纤维素(结构单元为D-葡萄糖)同样具有疏水性,但配位能力不及壳聚糖,加入纤维素后产物CO的选择性为9.1%. 上述对比实验结果说明,只有疏水性的添加剂与MPA-CdSe QDs表面Cd2+有效配位后才有利于光催化性能的提升. MPA-CdSe@chitosan组装体中较为稳定的Cd—N键使壳聚糖将量子点包裹于其疏水环境中,使MPA-CdSe@chitosan组装体整体表现出疏水性[28,29,40].
向纯水中加入一定质量的壳聚糖并测试溶液中CO2的溶解量,结果如图12 所示. 与纯水样品相比,壳聚糖(5.0 mg和10.0 mg)的加入使CO2的溶解量分别提高12.8%和28.2%,CO2的溶解量随壳聚糖加入量的增加而提高. 壳聚糖中丰富的氨基与CO2反应生成氨基甲酸盐是提高水中CO2溶解量的主要原因[22,25~27].
如前所述,关于CdSe 或CdS QDs 光催化还原CO2为CO 的中间体目前仍无定论[13,14],但是大家一致认为在光催化还原过程中,CO2首先在CdSe 或CdS QDs 表面吸附形成中间体. 在光照下,MPA-CdSe@chitosan 组装体中的QDs 受光激发产生光生电子-空穴对,电子跃迁至导带,空穴留在价带中. QDs导带电子还原吸附在QDs表面的CO2活性中间体最终将其转化为CO并释放;抗坏血酸钠捕获QDs价带中的空穴(Scheme 1). 壳聚糖的氨基与QDs 表面金属离子配位,将其包裹于壳聚糖的聚合物结构中,为QDs 提供疏水的微环境. 同时,壳聚糖的氨基与CO2可逆地生成氨基甲酸盐,提高了CO2在溶液中的溶解度并提高了QDs周围CO2的局部浓度. MPA-CdSe@chitosan组装体的这些特征共同促进了CdSe QDs在水溶液中光催化CO2还原效率和选择性的提升.
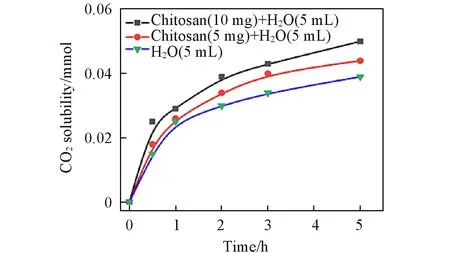
Fig.12 Effect of chitosan on CO2 solubility in H2O
3 结 论
在MPA-CdSe@chitosan 组装体中,壳聚糖通过氨基与Cd2+配位将量子点包裹于壳聚糖的疏水结构中. 在以NaHA 为电子牺牲体的条件下,MPA-CdSe@chitosan 组装体水相光催化CO2还原生成CO 的效率为73.6 mmol/g,选择性可达51.0%(光照60 h),相对于纯MPA-CdSe QDs,CO2还原反应的效率和选择性得到大幅提升,且表现出良好的稳定性. 光催化过程中,量子点的粒径逐渐增大,出现明显聚集,使量子点比表面积减小,表面活性位点减少,这是造成MPA-CdSe@chitosan 组装体光催化失活的主要原因. MPA-CdSe@chitosan组装体提升水相光催化CO2还原性能的原因主要有:(1)壳聚糖与量子点通过配位作用,将量子点包裹于壳聚糖的疏水结构中,为量子点提供疏水环境,抑制质子还原产氢;(2)壳聚糖提升CO2在水中的溶解度,为量子点提供富CO2环境.
猜你喜欢
大学物理(2022年9期)2022-09-28 01:10:52
现代畜牧科技(2021年9期)2021-10-13 06:38:40
粉末冶金技术(2021年3期)2021-07-28 06:26:50
物理通报(2020年7期)2020-07-01 09:28:02
童话世界(2017年29期)2017-12-16 07:59:32
中学生数理化·高二版(2016年6期)2016-05-14 13:19:33
精细石油化工(2015年3期)2015-12-14 09:07:42
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
应用化工(2014年11期)2014-08-16 15:59:13
