
CO2参与电化学构筑C—N键制备重要化学品
2022-08-06 04:38:26王茹涵贾顺涵吴丽敏孙晓甫韩布兴
高等学校化学学报 2022年7期
王茹涵,贾顺涵,吴丽敏,孙晓甫,韩布兴,3
(1.中国科学院胶体、界面与化学热力学重点实验室,中国科学院化学研究所,北京 100190;2.中国科学院大学化学科学学院,北京 100149;3.上海市绿色化学与化工过程绿色化重点实验室,华东师范大学化学与分子工程学院,上海 200062)
CO2是最主要的温室气体,同时也是重要的C1资源[1]. 截止到2022年5月,全球大气中CO2浓度已超过420 ppm(1 ppm=1 mL/m3,参见美国国家海洋和大气管理局数据https://gml.noaa.gov/ccgg/trends/).根据Manabe和Wetherald[2]共同开发的一维辐射对流模型发现,地表温度随CO2浓度升高而升高,大气中CO2浓度每增加1倍,全球平均温度将上升2.36 ℃. 自工业革命以来,人类过度依赖化石资源,导致过量的碳排放[3~6],尤其是化学制造业的CO2排放量占全球总排放量的近四分之一[7]. 因此,如何在现有能源系统的框架下解决好CO2排放问题具有重要意义. 电催化转化CO2近年来受到关注,该反应的主要产物包括CO、烃类、醇类和羧酸类等[8~12]. 与其它方法相比,电催化转化CO2条件可控,可以实现一些常规条件下难以进行的反应路径;其反应过程绿色、清洁. 可用水作氢源,显著减少了化学品的消耗和废液的产生. 此外,驱动催化过程的电能一方面可以来自于核能、风能、太阳能及潮汐能等清洁或可再生能源的发电,另一方面可以利用生产生活中非用电高峰期的“低谷电”,符合可持续发展的需要. 同时,电催化转化CO2合成重要化学品也是促进能源结构转型,实现碳中和的重要途径.
氮(N)元素是地球上最丰富的元素之一,以多种形式存在,包括氮气(N2)、有机胺、氮氧化物(如等形式,其中氮气主要占空气的78%,而硝酸盐、亚硝酸盐和有机氮化物是地下水、河流和湖泊的主要含氮污染物. 在影响地球的各种环境问题中,氮循环的人为扰动在短期内对人类健康和生物圈造成的影响更为严重. 其中,电化学N2还原反应(NRR)及硝酸盐还原反应(NITRR)成为温和条件下合成NH3的潜在途径[13,14].
将CO2和含氮物质(氮氧化物等)通过C—N构筑转化为高附加值化学品是一条有利于生态环境恢复的可持续发展策略. 但传统C—N键构建策略存在如下问题:碳源过度依赖于化石资源,面临全球能源紧缺及我国“双碳目标”发展等的多重挑战;现行的C—N键构建方式包括化石资源分离提纯、氨气合成、C—N键耦合等多个步骤,流程繁琐.
与传统热催化和其它加氢工艺相比,电化学法在原子利用及能源利用方面具有显著优势:(1)以温室气体CO2为碳源,降低对化石能源的依赖;(2)以大气中的N2或废水中的等(包括氮氧化物和有机氮源)为氮源,以H2O 作为反应过程中的质子来源,有效降低C—N 键耦合过程中的碳排放量;(3)使用耦合策略设计CO2与氮源共转化体系,实现一步法合成含氮有机化学品,简化生产流程及生产工艺,降低对生产设备的要求.
CO2(C=O键能为750 kJ/mol)和N2(N≡N键能为941 kJ/mol)等新型化学资源通常具有键能较高的惰性化学键. 这些惰性化学键的存在使得CO2与N2等分子参与化学反应时会出现化学反应热力学受限及反应动力学缓慢等问题. 因此,探寻惰性化学键的活化转化并探究其中的化学机制是实现通过CO2和N2等构筑C—N键合成含氮化学品的关键问题.
本文着重介绍了使用电化学策略,以CO2为原料构建C—N 键制备重要化学品的研究进展;并从反应过程、催化体系构建和机理探究的角度对尿素、酰胺(甲酰胺、乙酰胺等)、胺类化合物(甲胺、

Fig.1 Schematic illustration of CO2⁃involved electrochemical C—N coupling into value⁃added chemicals
乙胺、氨基甲酸酯等)的电化学合成进行了综述(图1). 重点讨论了CO2与不同氮源及有机胺类化合物)共同构筑C—N 键合成重要化学品的反应及机理. 同时针对目前相关研究中面临的挑战,对这一领域的发展做出了展望. 希望本文可以将惰性化学键的活化转化过程拓展到其它绿色碳资源(如生物质及其平台化合物、废弃塑料等),从而拓宽制备含C—N 键重要化学品的合成路径.
1 CO2参与电化学构筑C—N键的研究基础
CO2可以转化为高附加值化学品或燃料[15,16]. CO2的转化可以通过化学方法[17~20]、光催化[21,22]和电化学催化等方式完成. 虽然电催化CO2转化在可持续发展战略背景下具有良好的发展前景与应用潜力,但此过程中还存在产物选择性受限、副反应竞争、反应稳定性差、电流密度低和过电位高等缺陷.因此,研究人员从反应器、催化剂和电解液等方面开展了研究.
1.1 电化学体系构建
电催化CO2转化系统主要由电极、电解液及电解槽组成. 系统中的电极通常由粉末催化剂、聚合物黏结剂以及集电器组成. 但胶黏物会堵塞催化剂孔道,使传质通道受阻. 自组装及电化学沉积技术的不断发展也为电极的制备提供了新的途径[23]. 目前,关于水性、有机及离子液体(ILs)基电解液已有大量报道[24]. 水性电解液具有成本低、实用性强等优点,但水性电解液中CO2溶解度低、电位窗口狭窄等因素限制了CO2的反应效率. 有机电解液与水性电解液相比,CO2溶解度、电位窗口均有所改善,但其化学稳定性等问题也成为实际应用的壁垒[25]. ILs是近年来新兴的绿色溶剂,在气体吸收、材料制备及催化方面具有优势[26]. ILs具有低熔点盐的分子结构,使其有相对较高的CO2气体溶解度、较高的离子导电率及较宽的电位窗口. 在电化学构建C—N键过程中,常用中性无机盐溶液及ILs作为体系电解质,中性电解质溶液可以有效抑制析氢反应(Hydrogen evolution reaction,HER);而ILs一方面可以通过局部亲电和亲核区域的构建从而增强对底物的靶向吸附与活化,另一方面可以平衡HER,从而提高活性.
针对高效构建C—N键过程,电解槽的设计也是研究热点. H型电解池(以下简称H池)和流动电解池(以下简称为流动池)是CO2电化学构建C—N键过程中常用的反应器.
典型的H型电解池及流动池结构如图2所示. 其中,H池阳极室和阴极室由隔膜隔开,阴极室中包含工作电极(WE)及参比电极(RE),在阳极室中设置对电极(CE). 流动池同H型电解池结构相似,主要包含4个部分:支撑外板、流道隔板、气体扩散电极及离子交换膜[27],其中气体扩散电极和离子交换膜是流动池的两大核心部件.
气体扩散电极主要用于促进CO2向催化剂表面的扩散,即通过多孔疏水结构来促进催化剂表面形成气-液-固三相界面,从而促进反应的进行;离子交换膜则通过利用不同离子通道和离子传输规则将阴阳两极隔开,从而有效规避还原产物在阳极附近被氧化的风险. 离子交换膜主要分为阳离子交换膜(Cation exchange membrane,CEM)、阴离子交换膜(Anion exchange membrane,AEM)与双极膜(Bipolar membrane,BPM)3 类[28]. AEM 可以避免CEM 在高电流密度下阴极附近出现的酸化现象,从而有效推动CO2转化反应朝着生成产物方向进行,即有效抑制HER的发生,阴离子交换膜是电化学CO2转化反应中常用的离子交换膜. 双极性膜则可以将水溶液中电离出来的H+和OH‒分别输送到阴极和阳极,从而保持的阴阳两极pH 值的恒定,故可在一定程度上解决阴离子交换膜或阳离子交换膜存在的问题[29,30].
除了反应器各组件外,催化剂等也对电催化CO2转化反应具有直接影响. 电化学CO2转化反应中的电催化剂主要包括金属单质[31]、金属化合物(包括金属氧化物[32]、氮氧化物[33]和硫化物[34]等)、多孔有机框架材料[包括金属-有机骨架材料(Metal-organic framework,MOF)和共价有机框架(Covalentorganic framework,COF)等材料[35,36]]以及单原子催化剂[37,38]等(图2).
此外,电解体系中的电解质溶液的性质、pH值、离子种类等也会影响CO2转化的电流密度和产物的选择性等[39,40].

Fig.2 Electrolysis building blocks of CO2⁃involved C—N bond electrochemical construction
1.2 评价反应的参数
评价电化学催化反应过程中反应进行的程度与反应产物生成情况一般从以下几方面入手:(1)过电位:起始还原电位与平衡电位之差,其中起始还原电位是衡量催化剂性能的重要参数之一;(2)电流密度:衡量反应速率的重要参数,电流密度的大小受催化剂、电解质及电解池等因素的影响;(3)转化率:某一反应物的转化百分率;(4)法拉第效率:生成目标产物所消耗的电荷量占累计消耗电荷量的百分比;(5)稳定性:催化剂的活性及选择性随时间变化情况,一般用来衡量反应体系在电催化过程中的稳定程度;(6)产物生成速率:用产物生成速率来表示化学反应进行的快慢,受反应环境、催化剂等因素的影响.
由于构建C—N键反应过程中的反应活性较低,会受到多种环境因素的干扰,因此在探究C—N键生成机理时常采用对照实验的方式(图3),包括环境排除法及构建反应法. 环境排除法是指由于实验环境及化学品中有可能引入NOx,因而需要通过对照实验,进一步验证目标产物的生成的确依赖于原始底物,而非环境引入的污染物NOx;构建反应法是指通过构建新反应的方式探究C—N键生成机理.具体来说,反应实际上为中间体a与中间体b反应生成目标产物/目标产物前体c,现将假设中间体a′同已确认中间体b反应,在相同反应环境下,探究目标产物/目标产物前体c的生成情况. 若a′同b能够生成c,则可证明中间体a′为反应中间体a.
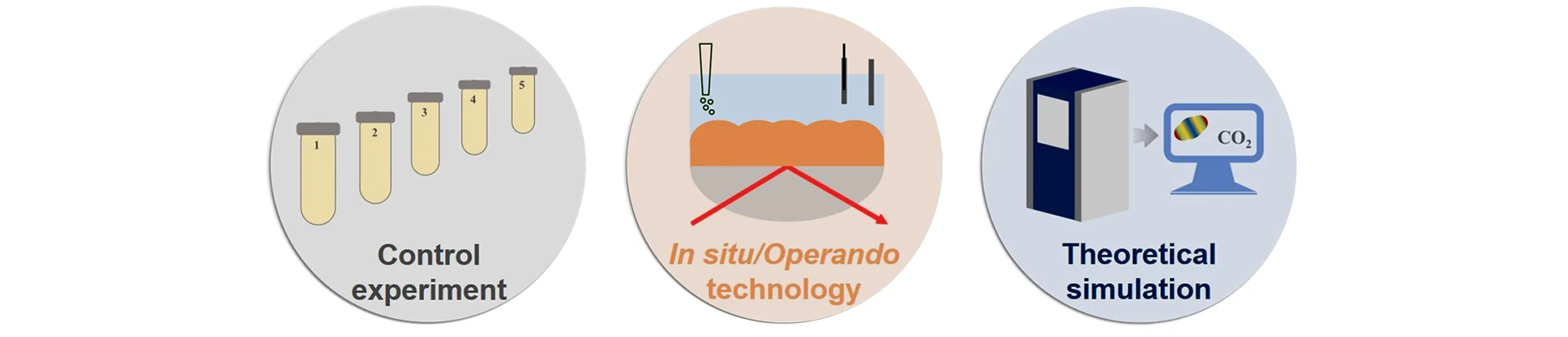
Fig.3 Methods for investigating the mechanism of electrocatalytic C—N bond construction with CO2
在探究C—N 键生成中涉及的关键中间体方面,由于反应过程中往往涉及到多种中间体,目前对于某种中间体是否参与成键耦合过程无法准确判断,通常在反应中采用显色反应(如,通过二乙醛一肟法与尿素产生的粉色溶液在525 nm 处的吸光度来证明溶液中尿素的生成)、气相色谱等方法对液相、气相产物进行定量;通过核磁共振波谱法(NMR)及比色法对电解质溶液进行分析从而对产物进行量化;通过同位素标记法对13C,15N等元素对C源、N源等进行追踪;同时多种原位表征及理论计算方法的综合利用对C—N键生成过程的探究也有重大意义,包括原位拉曼光谱、原位红外光谱、原位同步辐射技术、密度泛函理论(DFT)和程序升温脱附(TPD)等技术.
1.3 CO2和含氮小分子共电解
迄今,针对含氮小分子的活化转化,特别是电催化氮还原、硝酸根还原的体系[41,42]已经开展了大量研究. 而在电催化CO2还原的体系中,通过设计催化剂已经实现了CO2向CO、酸、醇的产品的高效转化[43~46]. 因此,将高反应效率的CO2转化与含氮小分子转化相结合,通过反应中间体C—N键的构筑形成“多组分共电解”策略,可达到生产高附加值产品的目的.
2 不同催化体系实现电化学构筑C—N键
近年来,电化学CO2还原反应过程中,C—N键的构建制备高附加值化学品成为热点. 目前,已经实现的主要反应过程如图4所示.
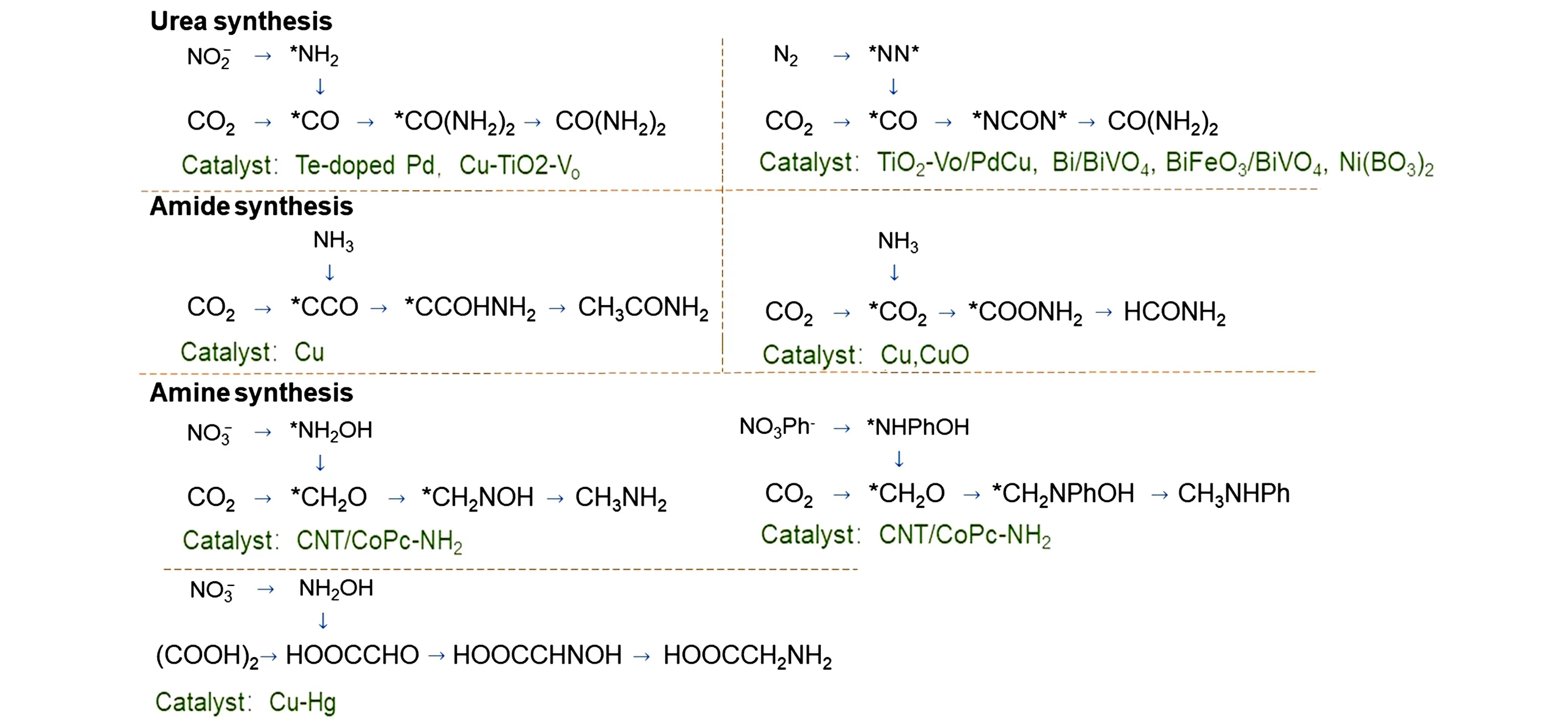
Fig.4 Synthesis of urea,amides and amines
2.1 无机氮源
2.1.1 N2作为氮源 气态氮元素是自然界中非常丰富的氮源,大气中约含有4000万亿吨气体,其中氮气占78%. 氮的利用主要以Haber-Bosch 工艺(400~500 °C,200~300 bar,1 bar=0.1 MPa)合成氨为主[47],由于N2中N≡N三键具有极高的键能(941 kJ/mol),因而该反应过程会伴随大量能源的消耗及大量CO2的排放(每合成1吨NH3将产生1.9吨CO2). 目前,通过Haber-Bosch工艺生产NH3所消耗的能耗约占全球能耗的1%~3%[48]. 考虑到绿色化学及碳中和等发展战略及目标,开发温和条件下高附加值化学产品的清洁生产技术势在必行. 而尿素作为氨的主要下游产物之一,是重要的化肥,也是一种重要有机化学品. 尿素的工业生产主要依赖于氨(NH3)和CO2采用Bosch-Meiser工艺在200 ℃及210 bar压力条件下,通过氨基甲酸铵(NH2COONH4)中间体,将NH3与CO2通过热化学偶联作用形成C—N键[47];胺和酰胺作为其拓展产物,主要是在100~250 ℃及50~250 bar压力下将NH3通过醇的胺化反应合成[49].
电催化转化技术是一种有望实现“零”碳排放的催化技术,电催化过程中所需要的能源可通过可再生电力提供,确保在常温常压条件下通过电化学氧化还原获得高附加值产品[50~52]. 电化学合成尿素的过程是气体不断消耗的过程,尿素的收率也容易受到气体特别是反应物低溶解度的限制. 因此催化体系的优化十分重要. 图2显示的H池需将气态原料首先溶解在水溶液中,通过扩散到电极表面参与反应. 具有气体扩散电极的流动池可以构建高效的三相反应界面,其中的疏水层阻挡了大部分水分子,只允许其中少量水分子通过作为合成尿素的质子源. 研究发现,在常温常压条件下,电化学催化N2与CO2可通过直接耦合的方式合成尿素[53].
Chen等[54]报告了使用PdCu-TiO2催化剂电催化合成尿素(图4),并对该过程中C—N键的形成机理进行了研究,在‒0.4 V(vs. RHE)的电压条件下,尿素的法拉第效率可达9.43%,产率可达3.76 mmol∙g‒1∙h‒1. 密度泛函理论(DFT)计算表明,吸附在催化剂表面的N2经活化后生成的*N=N*中间体与CO2在金属位点上还原生成的CO中间体经放热耦合后形成尿素前体*NCON*,经过连续的质子电子转移后还原生成尿素. 分别在H 型电解池和流动池中测试尿素的合成性能发现,在流动池中由于气体原料丰富,尿素的产率及法拉第效率与H型电解池中相比均有明显提高,同时也能够很好地抑制反应过程中的HER反应.
尿素合成过程中催化剂的优化设计还需关注气态分子的共吸附情况. Yuan 等[55]提出了一种Bi-BiVO4异质结构催化剂(图4),通过N2和CO2共活化合成尿素,在‒0.4 V(vs. RHE)电压条件下,尿素的产率可达5.91 mmol∙g‒1∙h‒1(0.29 mA/cm2),法拉第效率为12.55%. 催化剂吸附反应物后,*N=N*和*CO 中间体在反应决速步进行前耦合形成尿素前体*NCON*. 由程序升温脱附(TPD)测试结果可知,在催化剂Bi-BiVO4界面,不同电子与催化剂的亲和力不同,这也导致相关组分的空间电荷区域分布不均. 其中,KHCO3作为中性电解液,在NaBH4还原策略下使催化剂的BET比表面积大幅提升从而提高了催化性能,其比表面积从14.7 m2/g(BiVO4)提升至30.4 m2/g(Bi-BiVO4). 在对异质结构中分子吸附能力及局部电荷再分布情况进行改善后,Yuan等[56]将BiFeO3/BiVO4杂化物作为电催化剂(图4),该催化剂在‒0.4 V(vs. RHE)电压条件下实现了17.18%的尿素法拉第效率. 异质结处两种材料之间的接触导致电荷分布不均,从而促进了BiFeO3上CO2*活性中间体与BiVO4上*N=N*中间体的形成. 随后,他们[57]又通过一种简单的湿化学法制备了花瓣状Ni3(BO3)2纳米晶体,经退火处理后将水相合成过程中形成的Ni—OH键裂解,产生不饱和Ni位点,促进N2和CO2进行C—N键形成反应,在‒0.5 V(vs. RHE)电压条件下,尿素的法拉第效率可达20.36%(图4). 这是因为占据低轨道的Ni位点与相邻的表面羟基构成具有催化活性的受阻Lewis酸碱对,其可通过独特的“s轨道羰基化”实现对反应物的定向吸附和活化,并引发C—N的形成. 此外,他们[58]还设计了类似于米粒结构的InOOH纳米催化剂,该催化剂具有明确的Lewis 对活性位点,可以通过电子相互作用实现N2和CO2分子的定向化学吸附,在‒0.4 V 电压条件下,尿素的产率及法拉第效率可达6.85 mmol∙h‒1∙g‒1和20.97%. Yuan 等[59]通过设计一种新型的Co-PMDA-2-mblM(PMDA=均苯四甲酸二酐;2-mbIM=2-甲基苯并咪唑)导电MOF 材料,在‒0.5 V(vs.RHE)条件下尿素的产率达到14.47 mmol∙h‒1∙g‒1,法拉第效率高达48.97%,其中在CoO6八面体中产生了局域亲电与亲核区域,同时实现了Co3+的高自旋态向Co4+中间自旋态的转化,从而定向吸附N2和CO2分子,产生活性中间体*N=N*和*CO,进而有效触发C—N 偶联反应产生尿素前体*NCON*. Zhu 等[60]通过DFT计算,对二维金属硼化物(MBenes)上的电催化尿素合成过程进行了理论研究.
2.1.2 氮氧化物(NO-3/NO-2)作为氮源 在寻找氮气替代物的过程中,硝酸根离子脱颖而出,因为与N≡N三键(941 kJ/mol)相比,其中的键解离能(204 kJ/mol)相对较低,即转化过程中产物的反应动力学更有利[61]. 在工业生产过程中,大量含氮物质以亚硝酸盐和硝酸盐的形式排放到地表水中和的传统处理方式主要是通过催化将其转化为无污染的氮分子[62],虽然实现了对有害物质的无害化处理,但无法得到高附加值产品. 随后的研究集中在及电催化还原成氨,这可以解决上述下游产品制造的问题[63]. CO2与亚硝酸盐/硝酸盐的电催化偶联是解决上述问题的有效途径. 将原料从氮气拓展到硝酸盐领域,不仅可以实现CO2的资源化利用,还有助于控制水污染,实现尿素的高效绿色合成[64~67]. 最近,Lv等[68]通过前期*NO2与*CO2活性中间体直接耦合的方法实现了产物的高选择性生成. 尽管这种C—N偶联机制可以提高尿素的选择性,但是如何降低中间体*CO2NH2转化过程的高能垒也是未来需要探索的问题.
利用硝酸根与亚硝酸根作为氮源与CO2电催化合成尿素已经进行了不少研究. Feng等[65]使用Te掺杂的Pd纳米粒子[‒1.2 V(vs. RHE),5 mA/cm2]通过和CO2的共还原生成尿素(图4),其法拉第效率为12%. 理论计算结果表明,Te的掺杂可促进中间产物*CO的形成,即在CO2作为唯一反应物的情况下,通过Te掺杂Pd纳米粒子显著增加了反应过程中*CO中间产物的生成. 此外,Te掺杂还促进了形成*NH2的过程,且*CO和*NH2通过亲核攻击耦合的能垒也随着掺杂量的不同而改变. Meng等[69]利用具有氧空位的ZnO 作催化剂,将CO2和转化生成尿素,其法拉第效率可达23%[‒0.79 V(vs.RHE),5.3 mA/cm2]. 红外光谱测试结果显示,当CO2存在时,可以检测到最初假设的耦合中间体*COOH,但在电解液中加入后,该中间体消失. 这说明反应的关键步骤是中间产物*COOH和*NH2的耦合过程.
Feng等[65]和Meng等[69]将电合成尿素过程中涉及的机理解释如下:(1)与CO2中+4价态的C原子相比,*CO中间体处于还原状态,此后通过再次氧化的方式合成尿素;(2)*CO和CO在平衡条件下共存并可以相互转换. 据文献[70,71]报道,*CO可以通过2种方式引入体系:电化学CO2还原、直接引入CO气体(随后在Pd或Au等金属催化剂表面形成*CO);(3)反应中间体*NH2与*CO之间的反应可能是*NH2的亲核进攻. 因为CO2的亲电性比CO 更强,使得CO2更容易与亲核的含N 中间体发生偶联反应,此外*CONH2作为假设的稳定中间产物几乎不会受到*NH2的亲核进攻.
Saravanakumar等[72]使用TiO2/Nafion复合电极用CO2和作为反应物电化学合成尿素,尿素的法拉第效率可达40%[‒0.5 V(vs. RHE),0.8 mA/cm2]. Cao等[64]将负载Cu的TiO2纳米管作为催化剂,利用CO2和生成尿素,得到尿素的法拉第效率为43.1%[‒0.4 V(vs. RHE),3.2 mA/cm2]. 催化剂中TiO2富有氧空位有助于吸附NO-2并同时生成*NH2中间体. Lv 等[68]在In(OH)3催化剂上以和CO2为原料合成尿素,其法拉第效率可高达53%[‒0.6 V(vs. RHE),0.3 mA/cm2]. 他们[48]还在富有氧空位的氢氧化铟(Vo-InOOH)催化剂上,以CO2和硝酸盐为反应物电催化合成尿素. 研究表明,尿素合成过程中的决速步为*CO2NH2的质子化过程. 由于不饱和In原子上的In—O键先断裂,使得*CO2NH2质子化过程中的能量屏障降低,同时也提高了催化剂InOOH的电催化性能. 在0.1 mol/L KNO3介质中,催化剂Vo-InOOH 在‒0.5 V(vs. RHE)的电压条件下,尿素的平均产率可达法拉第效率可达51%.
酰胺在聚合物、医药等很多领域用途广泛,2019年Jouny等[27]报道了由CO和NH3生成酰胺的电化学C—N键生成反应(图4). 在300 mA/cm2的电流密度下,采用Cu颗粒作为电化学催化剂,可以实现产物乙酰胺的法拉第效率接近40%的选择性,反应中涉及CO 分子二聚化及表层水分子中H 原子的转移. CO水解后形成*C=C=O中间体,通过NH3对*C=C=O中间体的亲核进攻生成乙酰胺,同时抑制其它C2+产物的形成. 通过使用不同N 源可将这种电化学合成方式扩展到合成N-甲基乙酰胺(FE=42%)、N-乙基乙酰胺(FE=34%)及N,N-二甲基乙酰胺(FE=36%),该方法开辟了电化学生成酰胺的新思路.
Kim 等[73]利用作为氮源,将CO2电催化还原获得的草酸作为碳源,在Cu-Hg 电催化剂上合成甘氨酸(NH2CH2COOH)(图4),反应过程中甘氨酸的最大法拉第效率可达43%[‒1.2 V(vs. RHE),39 mA/cm2],甘氨酸的合成依赖于乙醛酸(HOOCCHO)和NH2OH 中间体,而Cu-Hg 催化剂又能有效解吸中间体,使两者能更容易发生偶联反应生成乙醛肟(HOOCCHNOH),进而在催化剂表面还原形成最终产物甘氨酸.
Li等[74]将CO2作为碳源,作为氮源,采用商用Cu或CuO颗粒作为电催化剂,构建C—N键形成甲酰胺和乙酰胺(图4),最大法拉第效率分别为0.4%和10%[‒0.58 V(vs. RHE)下,分电流密度分别为0.2和2.2 mA/cm2]. 通过机理研究发现,甲酰胺和甲酸酯的电合成过程中都利用到CO2经过活化生成的*CO2中间体,*CO2受到NH3的亲核进攻后,生成含C—N键的酰胺.
最近,Wu等[67]开发了一种由CO2和电合成甲胺(CH3NH2)的策略,采用钴β-四氨基酞菁分子非共价锚定在多壁碳纳米管上组成的多相催化剂(CoPc-NH2/CNT),实现了从CO2和电催化合成脂肪胺. 机理研究表明,级联催化过程中的关键步骤是甲醛(HCHO*)和羟胺(NH2OH)的自发缩合,两者分别是CO2还原为CH3OH及还原为NH3过程中的中间体. Tao等[75]报道了采用CO2和作为原料电化学合成乙胺的策略,反应在常温常压中性的水性电解质中进行,由氧化物衍生的铜纳米颗粒作为催化剂,乙醛肟(HOOCCHNOH)作为乙胺合成过程中的关键中间体,由乙醛(CO2还原为乙醇过程中的中间体)和还原为NH3的活性中间体)缩合反应形成.
2.2 有机氮源
2.2.1 有机胺作为氮源 Rooney等[76]报告了一种用CO2对胺、肼(N2H4)、羟胺和硝基化合物进行N-甲基化还原的电化学方法(图5). 该反应在水性介质中进行,并由多相CoPc基电催化剂催化,采用一锅法生成N-甲基胺,即,CO2经还原后生成亲电产物HCHO*,HCHO*与N亲电试剂经化学缩合还原生成最终产物. 同时,通过监测CH3OH与哌啶反应后对产物N-甲基吡咯烷酮(NMP)的选择性关系时,发现随着哌啶的在反应中浓度的增加,产物的选择性会有所变化. 在Mayr 等[77]定义的亲和性参数绘制的N-甲基化胺法拉第效率与CH3OH法拉第效率比率中,亲核性较高的胺能更有效地捕获HCHO*中间体,这可能与缩合步骤动力学有关.
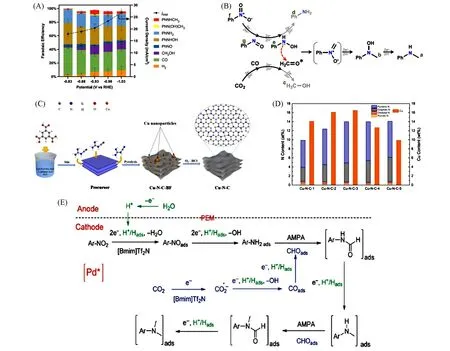
Fig.5 N⁃containing organic compounds as nitrogen source in C—N bond coupling
Feroci 等[78]报道了CO2与胺在离子液体中电化学合成有机氨基甲酸酯,目标产物的最高产率可达80%. Li等[79]利用苯胺和CO2合成N-苯氨基甲酸甲酯,Cu-N-C催化剂可实现产物52%的产率. 在催化剂制备过程中采用MOF衍生法(图5),在N掺杂的碳纳米片上合成原子分散的Cu-N-C材料,替代了用于电催化合成氨基甲酸酯的传统金属电极,这也使得催化剂具有更加丰富的活性中心,从而比传统金属电极具有更好的催化性能. Xiong 等[80]采用了一种绿色环保合成氨基甲酸酯化合物的方法,他们使用胺和N-烯基磺酰胺作为底物,在电化学催化条件下与CO2反应,从而合成氨基甲酸酯. 在探究过程中采用N-(2-乙烯基苯基)甲苯磺酰胺、吡咯烷和CO2作为模型底物,目标产物的产率可达77%. 氨基甲酸酯是天然产物和药物的重要中间体,通常在高压条件下利用CO2和胺形成的氨基甲酸酯阴离子,通过过渡金属催化或光催化与其它组分发生偶联反应.
2.2.2 硝基化合物作为氮源 Sun 等[81]提出了一种以H2O 作为氢源,通过CO2与硝基苯或其衍生物合成N,N-二甲基苯胺的策略(图5). 以Pd/Co-N/carbon 为电催化剂,氨甲基膦酸(AMPA)作为热催化剂,在常温常压条件下通过协同作用促进N,N-二甲基化反应的进行. 通过电解液中有无AMPA 的对照实验研究发现,AMPA的存在可以有效促进N,N-二甲基苯胺的生成,无AMPA存在时,CO2与硝基苯电化学反应只生成苯胺. 通过核磁共振波谱法(NMR)探究了AMPA协同作用机理,即:AMPA作为Brønsted碱可以激活苯胺上的质子,从而促进N,N-二甲基化的进行. 反应中,在AMPA 存在下,使用含有0.5 mol/L 1-丁基-3-甲基咪唑鎓双(三氟甲基磺酰基)酰亚胺([Bmim]Tf2N)的乙腈溶液作为电解质,发现在施加电位为‒2.3 V(vs. Ag/Ag+)时,硝基苯可完全转化,此时N,N-二甲基苯胺的产率可达92%. 对比不同Pd粒径的Pd/Co-N/carbon催化剂,发现可以通过粒径大小调节其电催化性能[82],从而调节反应速率及硝基苯的转化率.
3 总结与展望
本文综述了近年来CO2参与电化学构筑C—N 键制备重要化学品的研究进展,总结了不同氮源参与构筑C—N键的过程及其反应机理. 通过电催化C—N键生成反应合成尿素、酰胺、胺等重要化学品是从绿色化学及可持续发展角度出发的合成策略,这种方法不但与电催化CO2还原的优势相结合,同时也能够将反应体系拓展,并且采用不同的氮源. 通过对催化剂及催化体系的优化,已经实现了多个催化反应. 但电化学构筑C—N键过程中仍存在缺陷,如底物在反应过程中的中间体仍不能确定、与中间产物自还原相比C—N键构筑并没有体现出优越的竞争优势、催化剂在反应过程中同中间体的结合强度不高及产物选择性受限等问题,这些问题有望从以下3 个方面得到进一步解决(图6),CO2参与C—N 键的构建也有望得到进一步发展.
(1)构建多功能催化剂. 通过增加催化剂反应活性位点等策略,实现不同底物活化与转化. 由于CO2及含氮化合物在活化过程中涉及到多步反应及多电子转移过程,产生的中间体种类较多,例如CO2活化产生*CO,*COOH及活化产生*NH2,等,因此通过催化剂中多位点的协同作用,可以有效调控催化剂与反应中间体的结合方式及强度,从而提高反应活性及选择性.
(2)综合利用原位技术,将电催化C—N键生成反应中的反应机制及关键中间体精细化. 但在实际测试过程中,必须确保传质及反应环境等相关因素一致. 因为反应过程中需要将反应器与表征仪器相连,从而导致测试过程难度很大[83]. 未来利用原位拉曼光谱、红外光谱、X射线吸收光谱等技术同理论计算相结合,同时可与合成过程中的子反应(氮还原、亚硝酸盐/硝酸盐还原等)的反应途径、产物选择及催化剂性能等作参照对比,有望对C—N 键形成过程中和反应机理取得深入认识. 最近,Operando Raman光谱已应用于气体扩散电极CO2还原体系中,但还未应用于C—N耦合中[84,85].
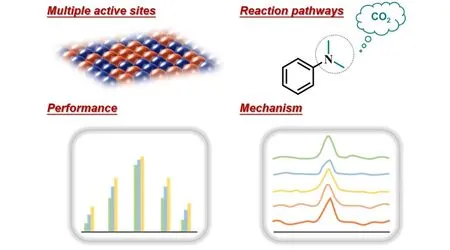
Fig.6 Proposed future directions of electrochemical C—N bond synthesis with CO2
(3)扩展电催化CO2合成化学品的范围. 现阶段电催化CO2构筑C—N键生成反应合成的含氮有机化学品主要包括尿素、酰胺(以乙酰胺为主)、胺(甲胺、乙胺及其衍生物)等,更多的含氮化学品有望通过电合成的手段获得,包括一些热化学催化难以或无法得到的化学品.
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
中国酿造(2016年12期)2016-03-01 03:08:11
中国资源综合利用(2016年7期)2016-02-03 03:00:13
中国酿造(2014年9期)2014-03-11 20:21:03
食品工业科技(2014年9期)2014-03-11 18:15:28
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年3期)2014-02-28 17:30:48
