
一锅三步由硝基芳烃合成邻氨基酚衍生物
2022-08-01 06:56孙承博张珠宝陈艺中匡禹豪廖小建冯鹏举
大学化学 2022年5期
孙承博,张珠宝,陈艺中,匡禹豪,廖小建,冯鹏举
暨南大学化学与材料学院,广州 510632
官能团及其转化是有机化学最基础的知识点,通过实验了解官能团的反应性质是掌握相关知识最直接的手段。将常见官能团的选择性连续转化融入一个紧凑的反应中,则便于学生直观学习。本设计实验的开展,有助于学生获得如下训练:第一,通过对现象的观测、结果的分析,使实验者了解各官能团的联系以及反应选择性的控制和实现;第二,通过紧凑多步的实验设计,使实验者实验时更专注于试验本身,减少以往有机反应不必要的实验等待过程,促使实验者统筹规划实验进行,做到有条不紊;第三,通过原位三步反应加料时间和方式的掌握,使学生了解观察实验现象的重要性以及反应时间对实验结果的影响;第四,硝基是涉及转化反应较丰富的官能团,通过该反应学习加深对基本官能团的认识,了解硝基转化产物的监测方法和产物数据表征;最后,通过该实验,使学生具备学科前沿研究的基本技能,创新性利用所学知识解决问题,并培养良好的科研素养(图1)。
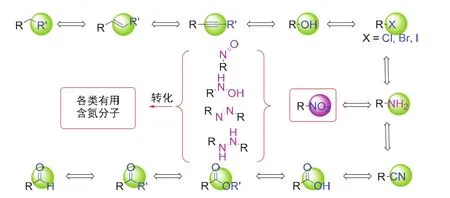
图1 官能团转化及硝基转化多样性示例
邻氨基酚类化合物广泛地存在于药物分子中,如GSK2126458,一种高效的口服磷脂酰肌醇III激酶抑制剂(PI3K),再如BMS-488043是治疗HIV感染的特殊小分子[1]。尽管这类分子展现出非常出色的药用价值,但关于这类分子实用的合成策略非常少见[2]。最早使用的合成方法是醇与各类缺电子芳基化合物发生SNAr型取代反应,而后再经硝基还原实现[3];金属催化的氨化及烷氧基化反应是合成此类化合物的常用方法,然而此方法的开展常需要对原料进行预官能团化,且反应中官能团兼容性较差[4]。N-芳基-O-保护的羟胺衍生物经[3,3]-σ重排是一个比较便捷的得到邻氨基酚类衍生物的途径[5,6],少数一些报道展示了此方法的高效性[7]。这类重排反应的实现依赖于原料芳基羟胺化合物的高效合成,通常羟胺化合物久置空气中容易氧化[8],同时芳基羟胺由硝基芳烃选择性还原得到,一般产物中会夹杂芳胺等副产物,分离十分困难[9],大大限制了此方法的应用。
利用[3,3]-σ重排由芳基羟胺衍生物获得邻氨基酚衍生物最关键的是如何高效、高选择性地实现硝基转化为羟胺以及羟胺的选择性保护。我们尝试以稳定易得的芳基硝化物为原料,将几步反应在一锅内实现,避免中间体的分离和保存。众所周知,官能团选择性转化的实现,需要精确选择反应试剂、掌控反应时间、调整加料策略。本实验选择活性较高的铑碳金属负载型催化剂,利用水合肼原位产生氢气为还原剂,实现高效、高选择性还原芳基硝化物为芳基羟胺,而后滴加乙酰氯的四氢呋喃溶液以及控制反应时间,实现羟胺的选择性保护,最后在三乙胺为碱的环境下,加入对甲苯磺酰氯实现[3,3]-σ重排,得到邻氨基酚衍生物(图2)。反应过程用TLC板实时监测,最终产物用1H NMR、13C NMR以及高分辨质谱加以确证。反应涉及多种官能团转化,现象丰富,反应操作多样,实验时间紧凑,适合培养学生观察和动手能力,同时帮助学生架构各官能团转化的知识网络。
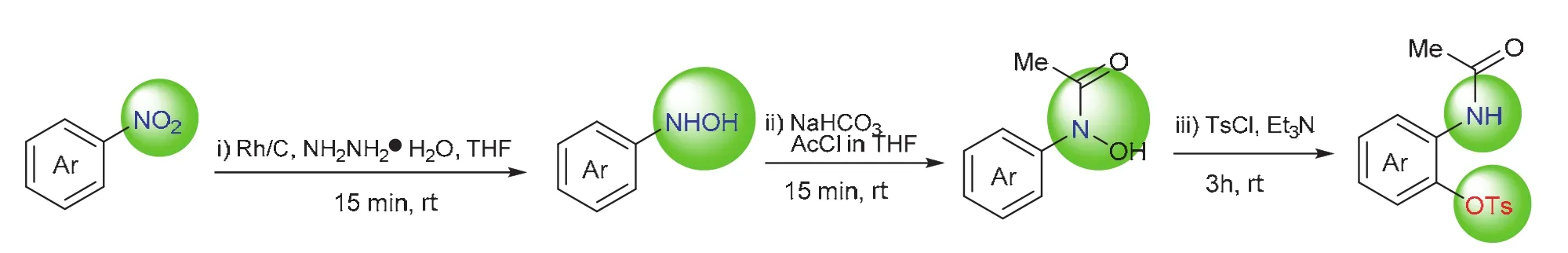
图2 一锅法实现芳基硝化物的多步转化
1 实验部分
1.1 实验原理
1.1.1 芳基硝化物还原为羟胺
硝基化合物的还原是一个复杂的过程,常常会生成多种副产物,如亚硝基化合物、羟胺化合物、芳基氨化物、氧化偶氮化合物、偶氮化合物以及1,2-二烃基肼(图3)。在中性或弱酸性条件下,硝化物的还原中间体的羟胺化合物进一步还原的速度比较慢,这为高选择性得到此类化合物提供了可能。本实验以铑碳金属负载催化剂,分解水合肼产生氢气,在中性条件下快速还原硝基化合物为芳基羟胺。

图3 硝基化合物可能的还原产物
1.1.2 芳基羟胺化合物的选择性保护
羟胺化合物有两个活性中心,即羟胺N/O亲核位点。当进行乙酰基保护时,由于N原子亲核性较强,优先被保护;若反应在较强碱性条件下(如三乙胺),则羟基会被夺去氢原子,随之进攻保护试剂乙酰氯,得到双保护产物;而选择性N保护中间体在反应体系中长时间不处理,其自身会发生N、O之间的乙酰基交换,而生成只有O-乙酰基保护的副产物(图4)。该步骤需要控制反应中选取的碱以去除生成的盐酸而不易引发副反应,同时要检测产物生成后,立刻进行反应后处理,避免重排反应发生。
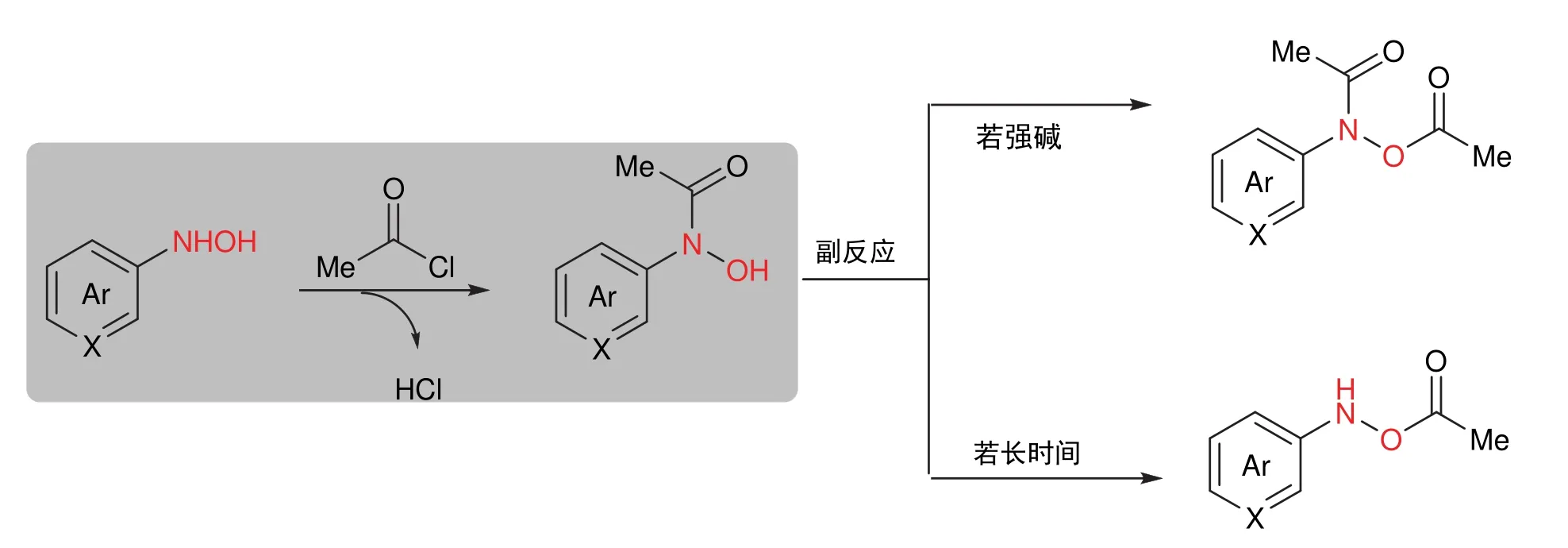
图4 羟胺化合物的选择性保护
1.1.3 [3,3]-σ重排
羟胺选择性保护的中间体在较强碱(三乙胺)的作用下与对甲苯磺酰氯反应,中间产物在室温下就可以经[3,3]-σ重排获得相应的邻氨基酚衍生物(图5)。
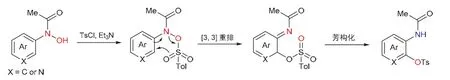
图5 [3,3]-σ重排反应机理
1.2 主要仪器和试剂
1.2.1 仪器
磁力搅拌器(MS5,Lab Fish)、循环水式多用真空泵(SHZ-D III,上海仪昕科学仪器有限公司)、旋转蒸发仪(RV 3 eco,IKA)、三用紫外线分析仪(ZF-6,上海嘉鹏科技有限公司)、电热鼓风干燥箱(DGX-9053BC-1,上海福玛实验设备有限公司)、核磁共振波谱仪(NMR) (Bruker Avance 300/400 MHz,以DMSO-d6/CDCl3为溶剂,TMS作内标,瑞士布鲁克科技有限公司)、高分辨液质联用仪(1290LC-6545 QTOF MS,美国安捷伦科技有限公司)。
1.2.2 试剂
5% Rh on Carbon (Rh/C) (Dry,阿法埃莎化学有限公司)、碳酸氢钠(NaHCO3) (AR,西陇科学股份有限公司)、乙酰氯(AcCl) (98%,安耐吉化学有限公司)、四氢呋喃(THF) (99.5%,安耐吉化学有限公司)、硝基苯(AR,天津市福晨化学试剂厂)、对溴硝基苯(AR,天津市福晨化学试剂厂)、对硝基苯甲酸甲酯(AR,天津市福晨化学试剂厂)、2-溴-5-硝基吡啶(AR,上海达瑞精细化学品有限公司)、2-氯-5-硝基吡啶(AR,上海达瑞精细化学品有限公司)、2-氯-3-溴-5-硝基吡啶和2-氯-3-甲基-5-硝基吡啶(AR,上海迈瑞尔化学技术有限公司)、水合肼(NH2NH2H2O) (98%,阿拉丁试剂上海有限公司)、三乙胺(Et3N) (AR,天津市大茂化学试剂厂)、对甲苯磺酰氯(TsCl) (99%,安耐吉化学有限公司)、乙酸乙酯(EtOAc) (AR,General-Reagent)、石油醚(PE) (AR,General-Reagent)、N-2-吡咯-5-硝基吡啶(> 95%NMR纯度,实验室自制)、N-2-吡唑-5-硝基吡啶(> 95% NMR纯度,实验室自制)、N-2-引唑-5-硝基吡啶(> 95% NMR纯度,实验室自制)。
1.3 实验步骤/方法
1.3.1 反应尝试及条件优化
我们选用实验室比较常用的四氢呋喃为溶剂,通过优化反应时间、酰化试剂的使用、加料时机的掌控,获得了最优的反应条件,具体实验条件的筛选和注意事项的声明如下:
(1) 尝试无水四氢呋喃和普通试剂级四氢呋喃,发现未干燥的四氢呋喃会大大降低铑碳的反应活性,反应中会有大量硝基化合物剩余,如果延长反应时间,则硝基化合物会绝大部分生成过还原产物芳胺。
(2) 尝试用含水的铑碳负载型催化剂,发现无法引发该反应。
(3) 尝试以乙酸酐代替乙酰氯进行酰基化反应,发现乙酸酐反应活性较低,需要较长的反应时间,从而使反应生成副产物。
(4) 反应中我们尝试各种碱,发现第二步酰化反应用碳酸氢钠效果最好,第三步重排反应用三乙胺效果最好。
(5) 由于对甲基苯磺酰氯为固体,其在称量定量上有一定优势,但气味较大,我们尝试液体苯磺酰氯为替代品,发现其反应效率与对甲苯磺酰氯相当,且可以直接用针头通过体积定量抽取,避免气味污染空气。若实验室通风效果良好,建议使用对甲苯磺酰氯,避免使用针头,产生废弃物。
(6) 本实验选用市售的7种廉价芳基硝化物及自制3种芳基硝化物,反应均表现出较高的效率,反应具有普适性。
(7) 反应具有丰富的现象,如气体产生、铑碳催化剂形态的变化、溶液颜色的变化等;反应产物可用多种方法观测鉴别,如第一步羟胺产物具有紫外显色性、磷钼酸显色性、碘熏显色性,第二步产物具有紫外显色性、碘熏显色性,第三步反应具有紫外显色性、碘熏显色性等。
1.3.2 代表性具体步骤
(1) 反应操作(各步反应现象如图6)。

图6 反应的实时监控图示
第一阶段:
反应在室温、空气条件下进行。2-溴-5硝基吡啶(1.0 mmol, 1.0 equiv.),5% Rh/C (6.6 mg,x(Rh) =0.6%),四氢呋喃(10.0 mL, 0.1 mol·L-1),磁力搅拌子置于100 mL的圆底烧瓶中,混合溶液在磁力搅拌器上搅拌。水合肼(60.0 mg, 1.2 mmol, 1.2 equiv.)逐滴加入上述反应液中。可观察到反应立刻产生大量气泡,分散很好的Rh/C催化剂聚集成块状。反应在室温下搅拌0.25 h,发现Rh/C大量黏附于瓶底。
第二阶段:
向反应体系中加入NaHCO3(102 mg, 1.2 mmol, 1.2 equiv.),随后加入乙酰氯(1.2 mmol, 1.2 equiv.)的四氢呋喃溶液(2 mL, 0.6 mol·L-1)。反应在室温下搅拌0.25 h,可观察到粘附于瓶底的Rh/C为黑色结块,溶液颜色发生明显变化。
第三阶段:
对甲苯磺酰氯(380 mg, 2.0 equiv.)以及Et3N直接加入上述反应溶液中,混合液在室温下搅拌3 h。有机相直接用装有硅藻土的砂芯漏斗过滤,用乙酸乙酯洗涤,收集有机相。
(2) 产物纯化。
向有机相中加入硅胶,(一般投入难挥发性物质1 g需加6 mL硅胶),进行干法拌样。旋转蒸发仪旋干反应液,得到产物分散较好的硅胶粉,经硅胶层析柱分离,淋洗剂为石油醚(PE)和乙酸乙酯(EtOAc)(V(PE) :V(EtOAc) = 5/1至1/1),试管收集淋洗液,薄层色谱板确定产物分布,合并有产物的组分,浓缩至干,得到白色至黄色固体,称量计算收率,收集固体于小玻璃瓶并做好标记,保存于冰箱。
(3) 产物结构测定。
反应产物溶于氘代氯仿或氘代二甲亚砜,利用核磁共振波谱仪测定氢谱和碳谱,确定产物的纯度和结构,进一步用高分辨质谱仪确定产物的正确性。
2 结果与讨论
2.1 反应的可重复性验证
实验选取2-溴-5-硝基吡啶为原料,组织三位实验者独立各进行5次实验(15次独立实验),综合评价反应过程和效果,所得结果如图7和表1所示。反应过程均用薄层色谱板监测(展开剂为V(PE) :V(EtOAc) = 3/1或5/1),现象基本一致,产率能够很好地复现。
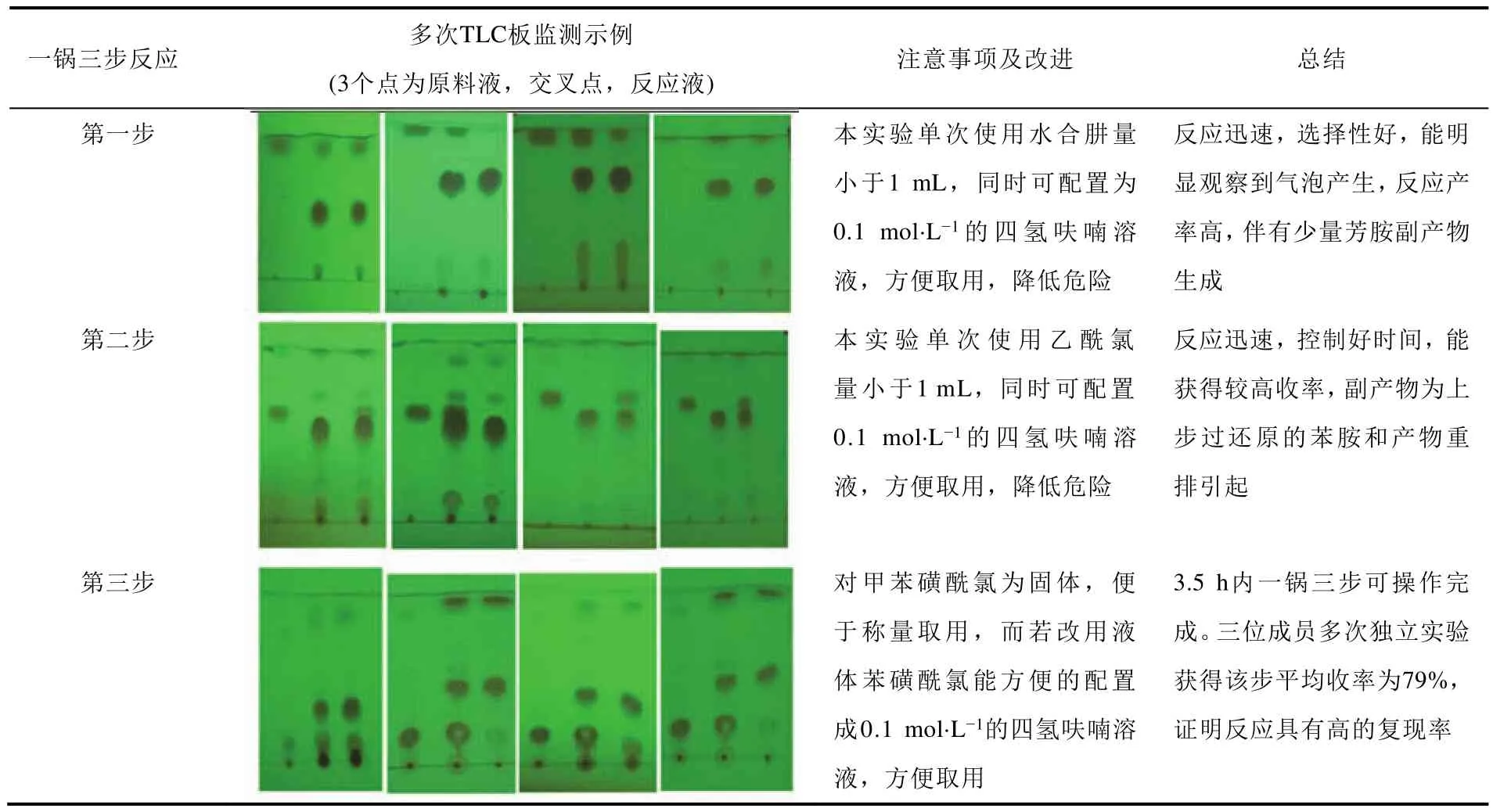
表1 反应的可复现性测试结果与分析
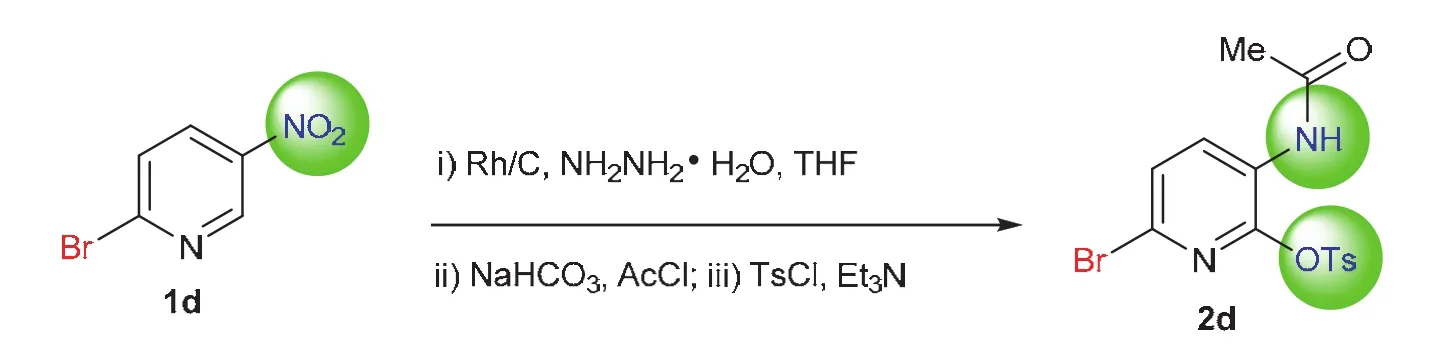
图7 反应方程式
2.2 反应的底物兼容性验证
得到模板反应良好的复现性,我们继续考察了该反应的底物兼容性。反应过程用TLC板实时跟踪,展开剂为石油醚和乙酸乙酯(V(PE) :V(EtOAc) = 1/1至5/1)。产物数据用NMR及高分辨质谱表征。结果表明反应具有良好的底物兼容性,取代的硝基苯以及3-硝基吡啶衍生物,均能在优化的条件下完成转化,产物均为白色至黄色固体,便于存放(图8)。产物具有多个官能团,便于后续衍生。
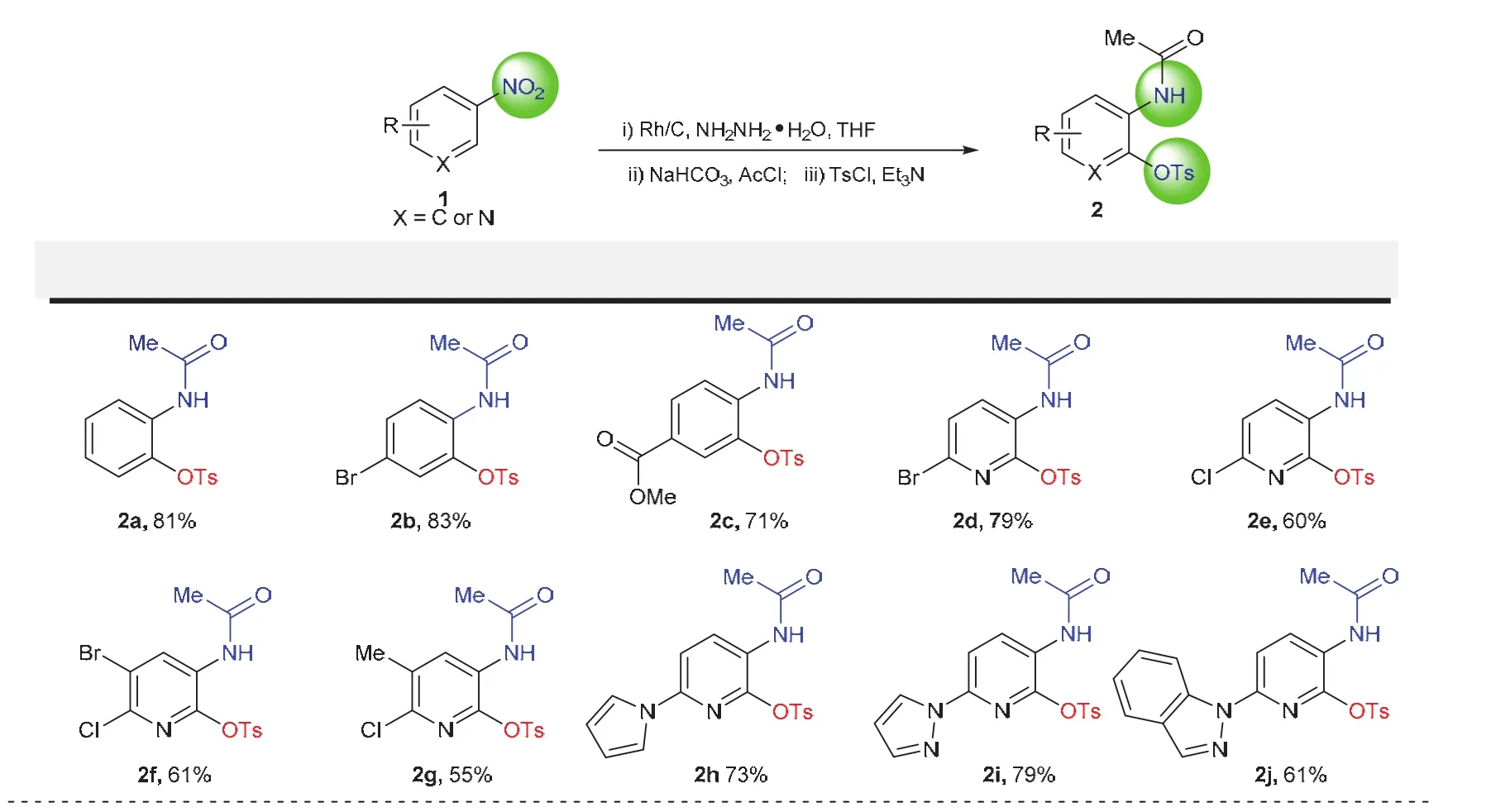
图8 反应的底物适用范围探索
2.3 反应特点、注意事项及反应实例
2.3.1 反应特点
该一锅法反应可以拆分成三步独立反应,每一步均能获得较高的产率,其中第一步和第二步反应后不用柱分离可直接用装有硅藻土的砂芯漏斗过滤,即可得到核磁纯度大于90%的产物。最终产物中的OTs基团可以在碱性条件下去保护获得羟基,乙酰基保护的芳胺在一定条件下脱除乙酰基,获得芳胺,同时邻位的氨基和羟基可继续衍生合成各类噁唑环。
2.3.2 反应注意事项
反应中使用了水合肼、硝基化合物、乙酰氯等,操作时应采用危险规避措施。学生操作时需要带手套和护目镜。反应中使用活性较高的铑碳催化剂,需要快速称量,同时其经过多步反应后活性降低,后处理危险性低。
2.3.3 反应实例
现以其中两个产物(2a和2f)合成为例,来展示化合物性状、反应效果和数据表征方式。
以硝基苯为原料经选择性还原、选择性羟胺保护及后续[3,3]-σ重排,以81%的收率获得了产物2a(图9)。反应分三个阶段进行,每个阶段我们均利用薄层色谱法进行监测,跑板情况通过紫外显色、磷钼酸显色以及碘显色观察,展示于图10。产物由核磁氢谱、碳谱及高分辨质谱表征,相关数据总结如下:2a为棕色固体,投入1.0 mmol原料可获取产物为247 mg,产率为81%,产物Rf= 0.30 (V(PE) :V(EtOAc) = 5 : 1);NMR数据:1H NMR (300 MHz, CDCl3, 25 °C,δ):8.22 (d,J= 8.2 Hz, 1H),7.75 (d,J= 8.4 Hz, 2H),7.57 (s, 1H),7.38 (d,J= 8.2 Hz, 2H),7.28 (d,J= 8.0 Hz, 1H),7.02 (d,J= 8.0 Hz,1H),6.90 (d,J= 8.0 Hz, 1H),2.52 (s, 3H), 2.05 (s, 3H)。13C NMR (75 MHz, CDCl3, 25 °C,δ):168.9,146.3,139.1,131.8,131.4,130.2,128.7,127.9,124.5,123.0,122.9,24.8,22.0。高分辨质谱数据:HRMS (ESI-TOF) (m/z),理论值C15H14NO4S-([M-H]-) 304.0649,实验值 304.0654。
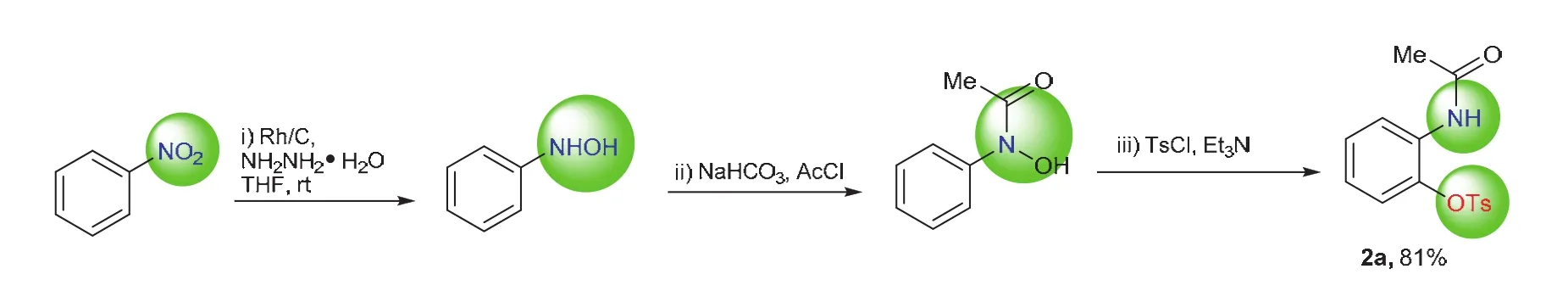
图9 化合物2a合成的反应式

图10 2a合成各阶段的TLC板监测图(原料,反应液和交叉点)
以2-氯-3-溴-5-硝基吡啶为原料经选择性还原、选择性羟胺保护及后续[3,3]-σ重排,以61%的收率获得了产物2f (图11)。反应分三个阶段进行,每个阶段均利用薄层色谱法进行监测,跑板情况通过紫外显色、磷钼酸显色以及碘显色观察,展示于图12。产物由核磁氢谱、碳谱及高分辨质谱表征,相关数据总结如下:2f为淡黄色固体,投入1.0 mmol原料可获取产物为255 mg,产率为61% , 产物Rf= 0.35(V(PE) :V(EtOAc) = 5 : 1);NMR数据:1H NMR (300 MHz, (CD3)2SO, 25 °C,δ):10.00 (s, 1H),8.70(s, 1H),7.87 (d,J= 8.3 Hz, 2H),7.50 (d,J= 8.3 Hz, 2H),2.43 (s, 3H),2.00 (s, 3H)。13C NMR (75 MHz,(CD3)2SO, 25 °C,δ):169.4,146.2,144.8,139.5,138.2,132.5,130.0,128.7,125.9,117.0,23.4,21.2。高分辨质谱数据:HRMS (ESI-TOF) (m/z),理论值 C14H12BrClN2O4SNa+([M +Na]+) 440.9282,实验值 440.9312。

图11 化合物2f合成的反应式

图12 2f合成各阶段的TLC板监测图(原料,反应液和交叉点)
3 结语
本实验结合本科有机化学的重点章节知识,将硝基化合物的多样性和选择性转化有机融入一锅法反应中,反应具有一定的底物普适性,反应产物具有多种官能团,便于后续衍生,且合成的邻氨基酚类物质在医药、染料等方面具有广泛应用。
本实验在室温条件下进行,操作简单,各步现象明显便于观察,产率较高,在较短时间内完成了硝基官能团的选择性还原、羟胺化合物的选择性保护以及室温条件下[3,3]-σ重排。该反应涉及多官能团的连续转化,便于学生对各类官能团建立关联性认知。
实验药品大部分为商业可得的原料,反应内容涉及TLC板反应监测,柱层析分离纯化产物,核磁测定反应产物纯度及结构,高分辨质谱确定产物分子量信息等,内容丰富。
本反应用时3.5 h左右,内容贴近课本基础知识,瞄准学科前沿,可操作性强,适合用于本科创新实验教学。
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
中学生数理化·高一版(2022年4期)2022-05-09
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
石油化工自动化(2020年1期)2020-03-05
通信技术(2019年8期)2019-09-03
中国循证儿科杂志(2019年2期)2019-06-04
浙江化工(2017年1期)2017-02-13
中学化学(2016年5期)2016-06-04
