
基于简化基因组测序技术的甘薯HRM分子标记开发及其应用
2022-06-30 08:50冯俊彦青莉芳屈会娟蒲志刚
核农学报 2022年7期
冯俊彦 郎 涛 张 聪 李 明 青莉芳 屈会娟蒲志刚,* 康 乐
(1 西华师范大学环境科学与工程学院,四川 南充 637002;2 四川省农业科学院生物技术核技术研究所,四川 成都 610061)
甘薯[Ipomoeabatatas(L.) Lam.]是世界上重要的粮食、饲料、工业原料作物[1]。在我国,甘薯是继水稻、小麦和玉米之后的第四大粮食作物,具有产量高、适应性广、抗逆性强等优点,对保障我国粮食与能源安全具有重要作用[2]。甘薯富含多糖、多酚等多种活性成分[3],具有重要的医疗保健作用[4],越来越受到消费者青睐。
甘薯是六倍体(2n=6X=90)作物,染色体多,基因组大,杂合度高,在基因定位、遗传图谱构建等研究上比其他作物困难[5]。目前甘薯遗传研究主要使用简单重复序列(simple sequence repeats,SSR)分子标记技术[6-7]、扩增片段长度多态性(amplified fragment length polymorphism,AFLP)分子标记技术[8-9]等,相关序列扩增多态性(sequence-related amplified polymorphism,SRAP)标记[10]、转座子插入标记(retrotransposon insertion polymorphisms, RIP)[11]、启动密码子靶向(start codon targeted polymorphism,SCoT)标记[12]等也有报道。与水稻、玉米等主要作物目前广泛使用的高通量单核苷酸多态性(single nucleotide polymorphism, SNP)分子标记技术相比,上述传统分子标记技术已经不能充分满足研究的需要。
近年来测序技术高速发展,给生物技术研究提供了强大的技术支持[13],其中简化基因组测序(reduced-representation genome sequencing, RRGS)技术是最具有应用前景的技术之一。该技术主要通过酶切技术降低物种基因组复杂程度,再针对基因组特定区域进行测序,从而获得基因组序列信息。目前该技术已发展出多种类型,主要包括特异位点扩增片段测序(specific-locus amplified fragment sequencing, SLAF)[14]、限制性酶切位点相关DNA测序(restriction-site associated DNA, RAD)[15-16]、基因分型测序(genotyping by sequencing, GBS)[17]等,其中应用较为广泛的是RAD测序技术。RAD测序技术具有高度的稳定性,可以获得大量的遗传多态性。与传统分子标记相比,RAD测序技术在发掘SNP方面具有显著优势,而且操作简单,试验费用低,对无参考基因组的物种也可以进行大规模SNP位点筛查[18]。目前RAD测序技术已成功应用于超高密度遗传图谱的构建、重要经济性状定位、群体遗传结构和系统演化分析等研究[15-16],并已成为甘薯遗传研究最理想的研究技术之一[16]。
目标SNP位点的准确分型是首先需要解决的问题。迄今已开发出杂交法、DNA测序法、微阵列芯片法、酶切法等20多种SNP分型方法[19]。相比其他方法,高分辨率溶解(high resolution melt, HRM)技术具有简便快速、通量高、成本低等优势。该技术是在PCR扩增产物变性过程中,实时检测体系内荧光信号的变化,实现对SNP位点进行基因分型[20]。在动植物基因分型、突变扫描等研究中已得到广泛应用[21-22]。
目前,关于甘薯单核苷酸多态性分子标记开发的研究较少[23-24],因此本研究通过生物信息学分析甘薯简化基因组测序数据,筛选稳定单核苷酸多态性位点,开发基于HRM技术的分子标记,旨在为SNP分子标记在甘薯研究中的开发及应用提供参考依据。
1 材料与方法
1.1 材料与试剂
本研究共收集国内外甘薯种质材料69份,其中国外种质材料7份、国内育成品种(系)49份、地方品种13份,全部试验材料均由四川省农业科学院生物技术核技术研究所收集保存(表1)。本研究使用的PCR试剂盒、DNA提取试剂盒、荧光染料均购自生工生物工程(上海)股份有限公司。
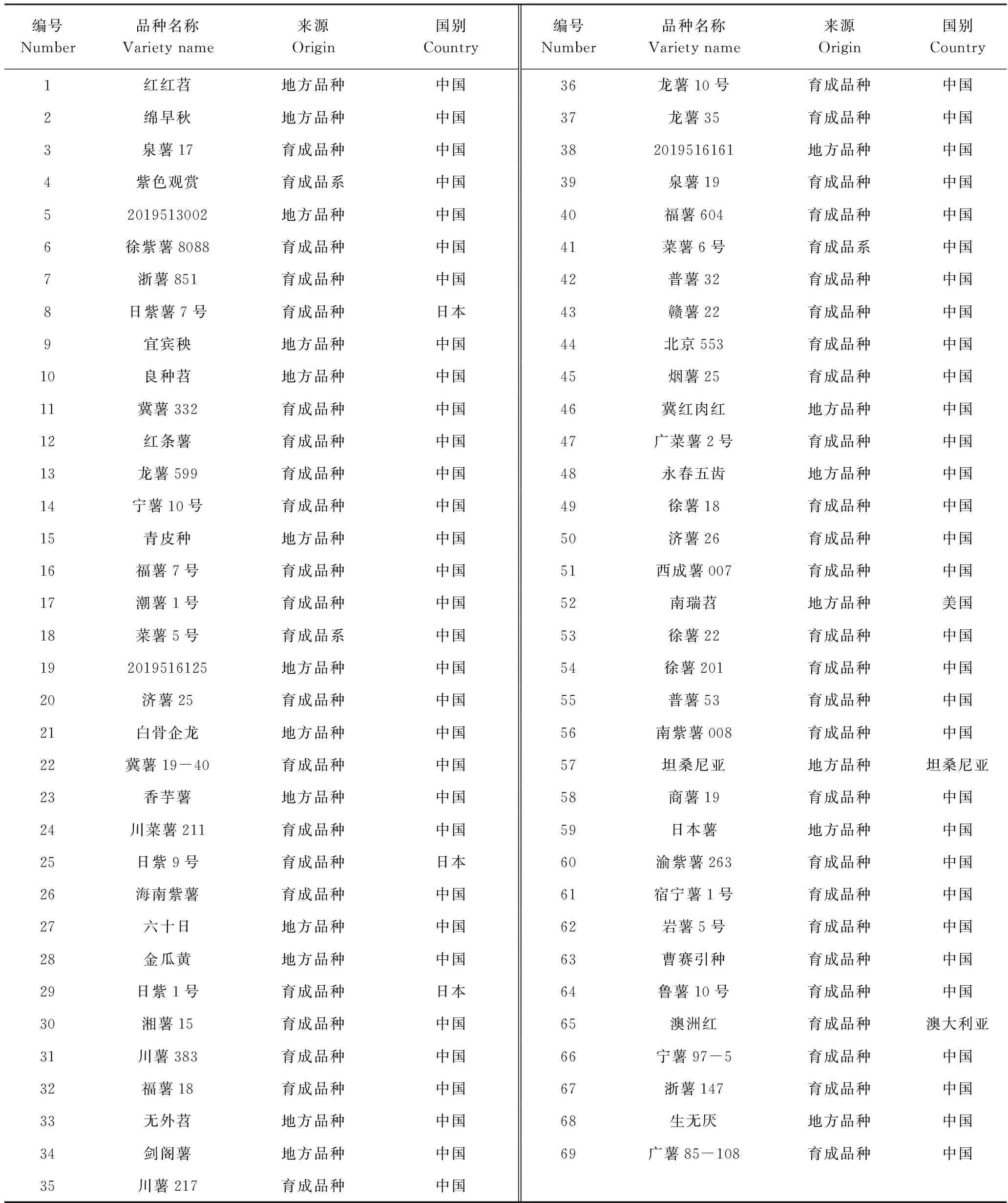
表1 甘薯种质材料信息Table 1 The information of sweetpotato accessions
1.2 仪器与设备
5425R微型离心机,德国Eppendorf公司;T100-PCR仪,美国Bio-Rad公司;NanoDrop One微量核酸蛋白浓度测定仪,美国Thermo公司;Hise 2500测序仪,美国Illumina公司;DYY-2C电泳仪,北京六一仪器厂;GelDocXR+凝胶成像系统,美国伯乐Bio-Rad公司;Lightscanner-Instrument 96高通量熔解曲线分析仪,美国Idaho-Technology公司。
1.3 试验方法
1.3.1 甘薯基因组DNA提取 2019年6月,将全部参试甘薯种质材料种植于四川省农业科学院生物技术核技术研究所新都试验基地。同年8月,选取各材料幼嫩叶片0.3 g,液氮冷却带回四川省农业科学院生物技术研究所生物技术育种工程中心,使用CTAB法提取各材料基因组DNA[25]。用超纯水充分溶解DNA后,抽取2 μL加5 μL上样缓冲液,用1%琼脂糖凝胶检测DNA质量。使用微量核酸蛋白浓度测定仪检测DNA浓度。根据DNA浓度,将DNA原液稀释到100 ng·μL-1,-20℃冷冻待用。
1.3.2 简化基因组测序及分析 根据RAD-Seq测序文库构建要求制备基因组DNA文库。先利用不同限制性内切酶对基因组DNA分别进行酶切,根据酶切试验结果选择限制性内切酶EcoRI和NlaIII对甘薯基因组DNA进行酶切。按照RAD简化基因组测序建库方法,构建长度范围在300~400 bp的双端测序(pair-end)文库。最后利用Ilumina HiSeq双端测序平台进行测序。
用Stacks v1.40软件过滤原始测序数据[26],去除接头序列、低质量测序数据、接头序列不明确的序列,以及长度小于指定长度的序列。选取过滤后测序片段长度的15%进行质量评估,保留质量高于10的序列。根据相同酶切位置测序片段之间的相似性,进行计数并获得每个接头序列的数目和深度信息。为保证后续获得准确的SNP位点,过滤掉接头序列中5′端非酶切位点为ATC起始的序列和深度值为1的序列,统计数目和深度。利用Stacks v1.40软件中的无参考基因组分析方法,将个体内的测序片段进行内部比对,生成各个材料的stacks文件,再对不同个体进行两两比对,比对时允许的错配数为2。整合深度信息和比对结果信息,过滤掉数据缺失大于30%的分型结果。利用最大似然法得到高可信度的SNP基因型结果[27]。
1.3.3 引物设计合成及PCR条件 根据含有高可信度SNP的测序片段序列信息,筛选SNP位点位于序列30~90区间内,且仅有1~2个SNP多态性位点的序列,使用BatchPrimer3 (http://batchprimer3.bioinformatics.ucdavis.edu/cgi-bin/batchprimer3/batchprimer3.cgi),设计特异PCR引物,由生工生物工程(上海)股份有限公司合成。
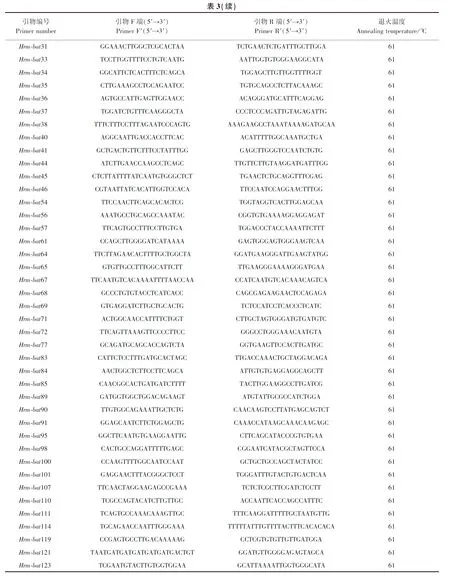
PCR反应体系10 μL:50 ng·μL-1植物基因组DNA 2 μL,10×buffer 1 μL,25 mmol·L-1MgCl20.6 μL,2.5 mmol·L-1dNTP 0.8 μL,10 μmol·L-1上、下游引物各加0.25 μL,5 unit·μL-1Taq酶0.1 μL,10×Evagreen荧光染料1.0 μL,10 μmol·L-1高、低温内标各加1.0 μL,最后加去离子水2.0 μL补齐。反应程序:94℃预变性5 min;94℃变性30 s,61℃退火30 s,72℃延伸45 s, 36个循环;72℃终延伸 min,慢慢冷却至12℃。
1.3.4 PCR扩增产物检测 将PCR反应板中的PCR产物转移8.5 μL至上样板中,每孔加入15 μL矿物油离心后,将上样板放入Lightscanner仪器中,进行检测。检测程序包括采集62~95℃之间荧光信号变化数据,升温速率设定为0.1℃·s-1。
PCR产物中加入5 μL上样缓冲液(40%蔗糖,0.025%溴酚蓝),用1.5%(w/v)的琼脂糖凝胶进行电泳分离。在荧光成像仪上检测拍照。
1.4 数据统计及处理
数据采集完成后,使用Lightscanner仪器自带的溶解曲线分析软件对结果进行分析。在分析过程中,根据仪器提示或手动剔除PCR无扩增的样本,高低温内标根据扫描结果手动设置,以样本曲线高、低温内标曲线峰的最高点作为高低温内标的温度点。然后分析各样本熔解曲线的分型结果。检测各样本分型检测,对个别未正确分型的样本,进行手动矫正。
利用Excel 2007软件对分型数据进行初步处理,并进行数据相应格式转化。利用SPSS 16.0软件的聚类分析模块,对参试材料进行聚类分析。
2 结果与分析
2.1 简化基因组测序结果分析
为尽可能多挖掘甘薯基因组中遗传多样性丰富的SNP位点,本研究根据多年田间表型鉴定数据以及品种来源信息,选取23个遗传多样性丰富的甘薯种质材料进行RAD简化基因组测序,共获得54.13 Gb数据,从中过滤掉序质量小于9的数据和不符合要求的数据后,得到样本测序数据的Q20值最大为96.26%,最小为93.44%,平均Q20值为95.42%。Q30值最大值为90.63%,最小为85.60%,平均Q30值为89.16%。
对23个甘薯品种原始测序数据过滤后,获得高质量测序数据38.18 Gb数据,平均每个样本获得1.66 Gb数据,共获得155 467 607个双端测序片段以及 7 216 825 个单端测序片段。共获得RAD标签 11 739 112 个,单个材料最大为612 312个,最小为369 969个,平均每个样本获得510 396个。共获得 155 751 596 个测序片段,平均每个样本获得6 771 809 个。通过计算每个样本中获得的总测序片段数与其总RAD标签数比值,获得测序深度,23份材料的测序深度处于9.87~16.44之间,平均测序深度为13.08。不同样本的原始数据分布范围为6 433 720~28 746 118,平均 10 541 122 (表2)。利用Stacks v1.40软件进一步过滤数据并对样本之间进行比较分析,发现346 609个片段含有多态性单核苷酸位点,共检测到835 756个SNP位点。

表2 甘薯种质材料的RAD简化基因组测序数据统计Table 2 The statistic raw reads of RAD sequencing of sweetpotato accessions
2.2 单核苷酸分子标记开发
为确保SNP数据的可靠性,以简化基因组测序数据分析获得的SNP数据为基础,对SNP进行再次严格过滤,确保单个SNP在95%以上的测序样本(≥22个)中被检测到,并对单个测序片段中出现3个以上的SNP位点的序列进行过滤,最后选取3 650个高质量测序片段。根据高分辨率熔解曲线技术对扩增产物片段在100 bp左右的要求,进一步对测序片段进行了筛选,只保留SNP位点位于序列36~90 bp范围仅有1~2个SNP多态性位点,且主要变异类型为A/C、A/G、T/C、T/G类型的序列,利用BatchPrimer3在线程序设计引物。针对1个单核苷酸多态性位点成功开发54对引物,针对2个单核苷酸多态性位点成功开发80对引物。为验证开发引物,选取8个甘薯品种DNA作为模板,使用高分辨率溶解曲线技术,分别对134对引物,扩增产物进行检测,结果表明,134对引物中,112对有扩增产物,占总引物的83.58%,22对引物没有扩增产物,占总引物的16.42%。在有扩增产物的134对引物中,15对(11.19%)对引物扩增产物检测结果为多峰,扩增产物不止一个,36对(26.87%)引物在8个样本中仅有一个扩增产物,样本之间没有差异,61对(45.52%)引物在8个样本中的扩增特异性好,仅有一个扩增产物,而且样本之间有差异,其中,27对引物之间差异较小,可以对小样本的不同基因型进行区分(表3)。34对引物扩增多态性丰富,差异显著。对扩增产物单一的引物,琼脂糖电泳验证结果表明,这些引物仅有1条100 bp左右的扩增产物,特异性良好(图1)。

表3 扩增多态性引物信息Table 3 The amplified polymorphic primer information
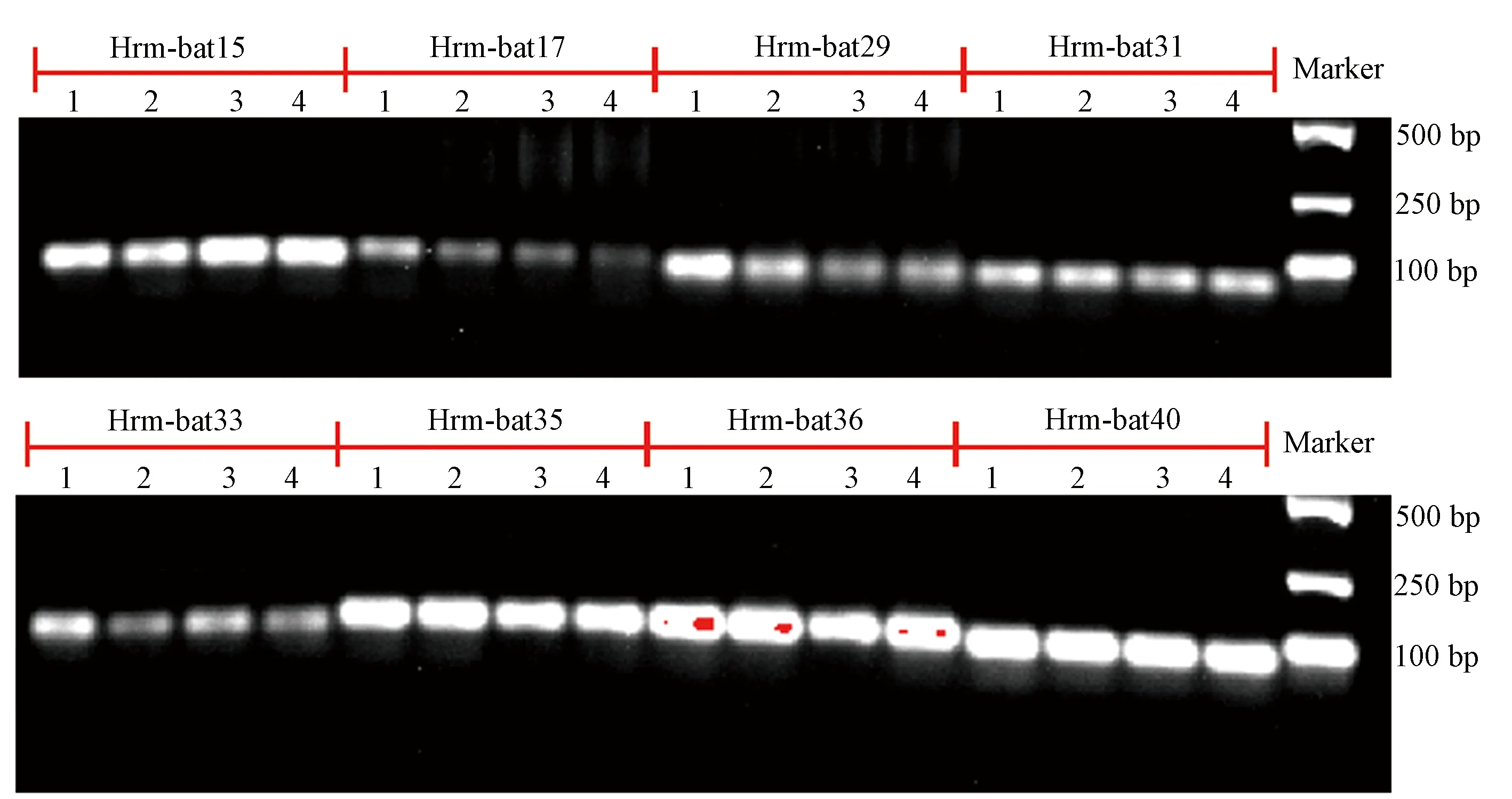
注:Marker:DNA Marker,标准条带分别为100、250、500 bp;1~4: 不同的参试材料。Note: Marker:DNA Marker. The size of bands is 100, 250, 500 bp, respectively. 1~4: Different sweetpotato accessions.图1 部分Hrm-bat引物在参试材料中扩增产物电泳图Fig.1 The electrophoretogram result amplified by primer Hrm-bat in different accessions
2.3 基于HRM检测技术的分子标记多态性分析
为进一步验证新开发引物在不同来源甘薯种质中的应用价值,在初步筛选的基础上,选取34对扩增产物单一,差异显著,且多态性丰富的引物,对本课题组近年来收集、保存且具有代表性的育成甘薯品种、地方甘薯品种、引进甘薯品种及部分测序甘薯品种共52份进行鉴定。结果表明,34对Hrm-bat引物在52份甘薯种质材料共检测到296个多态性位点,平均每对引物检测到8.71个多态性位点(图2)。引物Hrm-bat54、Hrm-bat69检测到的位点数最多,分别检测到14个多态性位点。引物Hrm-bat25、Hrm-bat89、Hrm-bat100、Hrm-bat107检测到的位点数最少,分别检测到5个多态性位点。根据检测结果筛选到14对引物检测到的10个以上多态性位点,分别是Hrm-bat15、Hrm-bat30、Hrm-bat45、Hrm-bat46、Hrm-bat54、Hrm-bat57、Hrm-bat61、Hrm-bat69、Hrm-bat72、Hrm-bat85、Hrm-bat90、Hrm-bat95、Hrm-bat111。
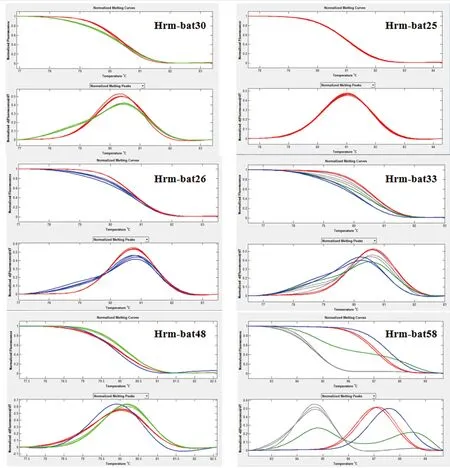
注:本图片为Lightscanner仪器自带软件分析结果。横坐标是温度值,纵坐标为标准化荧光信号值。Note:The picture shows the analysis results of the Lightscanner instrument’s software. The abscissa is the temperature and the ordinate is the normalized fluoresence.图2 部分Hrm-bat引物在8份甘薯材料中的HRM检测结果Fig.2 Applications of some Hrm-bat primers in 8 sweetpotato accesssions with HRM
为验证Hrm-bat分子标记在甘薯种质材料间的多态性,本研究对Hrm-bat分子标记在52份甘薯种质材料中的扫描结果进行材料间的比对分析(图3),结果表明,34个Hrm-bat分子标记在甘薯种质1-24和紫色观赏中获得的多态性位点最多,有30个位点具有多态性,多态性率达到90.91%。甘薯种质2019516161和川菜薯211之间的差异最小,仅得到9个多态性位点,多态性率只有27.27%。所有Hrm-bat在全部参试材料间的平均多态性达到59.35%。以上结果表明,本研究设计开发的Hrm-bat分子标记在甘薯种质材料间具有丰富的遗传多态性,可以用于发掘甘薯种质材料之间的遗传多样性研究。
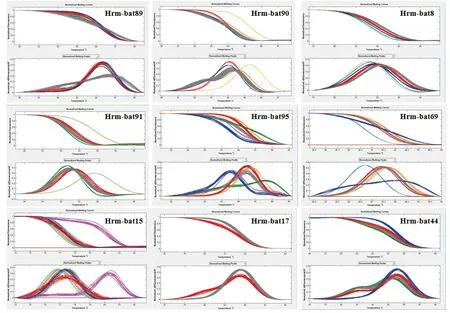
图3 部分Hrm-bat引物在52份参试材料中的HRM检测结果Fig.3 Applications of some Hrm-bat primers in 52 sweetpotato accessions with HRM
2.4 甘薯种质材料遗传多样性分析
基于34对Hrm-bat分子标记对52份参试甘薯种质材料的分子标记数据,利用SPASS V16.0软件计算不同材料间平方Euclidean距离,使用系统聚类法的组间联结法进行聚类分析。结果表明,52份参试材料可分为两大类,其中第一大类群包含51份甘薯材料(图4);另一大类群仅包括1份紫色观赏甘薯材料,该材料以台湾紫秧为母本,通过放任授粉选育得到。
在聚类图重新标定距离约5.5处,第一大类又被划分为5个小类群。其中,第一小类群最大,包含41份甘薯材料,占总材料的78.85%。该类群包括5个亚群,第一亚群包括5份材料,其中福薯18、北京553、冀红肉江88、广菜2号关系较近,香芋薯关系较远。第二亚群为最大群,包含18份材料,分为4组,4组之间关系较近,甘薯骨干亲本南瑞苕也在本类群中。第三亚群包括10份甘薯材料,分为两组,包含了甘薯骨干亲本徐薯18。第四亚群包括5份材料,其中,地方品种170和湘薯15之间、日紫9号和福薯604之间关系较近,冀19-40关系较远。3份材料(川薯383、良种苕、西城薯007)与其他材料关系较远,聚为一个亚群(图4)。

图4 基于Hrm-bat分子标记的52份甘薯种质材料平均连锁聚类图Fig.4 Dendrogram of 52 sweetpotato accessions using average linkage based on Hrm-bat molecular markers
第二小类群包含3份材料,分别是菜薯5号、金瓜黄11、2019513002。第三类群包括2份材料,分别是湘薯15号、无外苕。第四小类群包含红红苕、济薯26、赣薯22、绵早秋共4份材料,其中红红苕、济薯26、赣薯22关系较近,地方品种绵早秋关系较远。第五小类群包含日紫1号1份材料(图4)。从聚类图可知,日紫1号与其他材料之间的关系较远,可能与其国外来源有关。
整体来看,聚类图中参试52份甘薯种质材料之间遗传关系较近,大部分选育品种之间差异较小,这可能与我国甘薯育种中广泛使用少数材料作为亲本,选育出的品种间亲缘关系较近有关。相对而言,一些引进材料和地方品种的遗传差异较大,可作为育种亲本使用,从而进一步丰富品种的遗传多样性。
3 讨论
目前,甘薯研究中主要使用的分子标记仍然是非特异性、传统型分子标记[28-29]。相比传统分子标记,SNP分子标记在基因组中分布广泛,标记数量远多于传统型分子标记[30],但是目前该标记在甘薯研究应用较少。
测序技术的发展为甘薯研究提供了强大的技术支撑,甘薯及其近缘种I.trifida和I.triloba基因组测序工作相继完成[31-32]。在众多测序技术中,简化基因组测序技术在发掘基因组SNP多态性方面优势也逐渐显现[33],目前已开发出多种简化基因组测序技术,这些技术已经在甘薯超高密度遗传图谱的构建、群体遗传结构和系统演化分析等研究中得到广泛应用[34]。本研究利用简化基因组测序技术对甘薯种质材料进行了测序分析,在甘薯基因组中检测到835 756个SNP位点,从中筛选了一批多态性高、稳定好的SNP位点,用于开发SNP分子标记。同时,本研究将简化基因组测序技术应用到甘薯基因组SNP发掘和分子标记开发上,充分发挥了简化基因组测序技术高通量、快速、低成本的优势,为今后甘薯SNP分子标记的开发提供了参考。
近年来,SNP分子标记的检测技术一直是国内外研究的热点。目前,研究人员已开发出多种SNP检测技术。相比其他检测技术,HRM技术操作简单、灵敏高效,能够开展高通量、低成本的操作,可以有效区分SNP、SSR和InDel等不同类型变异的杂合与纯合状态,在水稻、玉米、小麦等作物的遗传及分子标记辅助选育研究上得到了广泛应用[20-22]。因此,本研究根据HRM技术要求设计开发引物,以简化基因组测序筛选到含有SNP多态性位点的测序片段为参考,成功开发出134对分子标记,通过对开发分子标记的验证,筛选到98对引物的扩增产物特异性好,利用HRM技术实现了对不同基因型的准确区分。
为进一步验证本研究开发的引物在甘薯种质材料中的多态性,本研究选取了34对多态性高的引物对52份甘薯种质材料进行了分型,发现平均每对引物检测到8.71个多态性位点,最多可以检测出15个基因型,最少仍可检测出5个基因型。初步证明本研究开发的HRM分子标记在甘薯种质材料中具有丰富的多态性,在今后甘薯分子标记研究中具有一定的应用价值。
利用本研究开发Hrm-bat分子标记的检测结果,对52份甘薯种质材料进行的聚类分析发现,52份材料件相似性很高,多数材料的差异在5.5以下,这与我国甘薯育种中使用亲本较少,育成品种遗传背景狭窄有密切关系,与前人研究结果基本一致[35-36]。78.85%的参试材料被聚到一个类群中,其中包含了甘薯骨干亲本南瑞苕和徐薯18,这可能与我国大量甘薯品种含有骨干亲本其血缘有关[37-38]。相比较而言,聚类图显示甘薯地方品种和国外引进品种的遗传差异较大。建议在今后甘薯育种中尽量选用地方品种和国外甘薯品种,以更好地拓宽甘薯遗传背景[39]。在聚类图中一些地方品种、国外引进品种与育成品种有聚集,推测这些品种间存在亲缘关系,具体原因还需进一步研究。
4 结论
本研究利用简化基因组测序技术对甘薯地方品种、引进品种及育成品种进行了测序分析,成功开发出基于HRM技术的甘薯特异SNP分子标记,并将部分分子标记应用于甘薯亲缘关系分析,为今后甘薯SNP分子标记开发提供了参考。
猜你喜欢
广西植物(2022年8期)2022-09-07
渔业科学进展(2022年4期)2022-09-05
中国农学通报(2022年12期)2022-06-01
中国典型病例大全(2022年9期)2022-04-19
中学生物学(2019年7期)2019-10-17
农民致富之友(2019年23期)2019-08-16
科技资讯(2016年32期)2017-03-31
世界热带农业信息(2016年3期)2016-04-05
小学生导刊(中年级)(2009年6期)2009-11-10
中国当代医药(2009年22期)2009-05-14
