
p38 MAPK抑制剂通过抑制NLRP3途径介导的细胞焦亡对小鼠慢性阻塞性肺疾病损伤的改善作用
2022-06-18 06:19王秋婷石慧芳
吉林大学学报(医学版) 2022年3期
李 明,王秋婷,陈 山,石慧芳
(海南医学院第二附属医院呼吸内科,海南 海口 570311)
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是一种临床常见的肺部疾病,并以不可逆性气流受限和持续性呼吸道症状(如黏液分泌过多)为主要特征[1]。吸烟被认为是造成COPD形成与发展的主要病因。研究[2]证实:吸烟不仅能够招募并活化炎症细胞(尤其是巨噬细胞和中性粒细胞),还能促进炎症因子如肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)和白细胞介素1β(interleukin-1β,IL-1β)的释放。香烟烟雾作为炎症信号的刺激因子,能够激活p38丝裂原活化蛋白激酶(mitgen-activated protein kinase,MAPK)和核因子κB(nuclear factor-κB,NF-κB)信号通路,加重呼吸道的炎症反应[2-3]。但迄今为止,香烟烟雾上述作用的具体致病机制尚不清楚。
巨噬细胞是免疫系统中的重要组成部分,在先天性免疫和获得性免疫中均发挥着不可缺少的作用。近年来有研究[4-5]显示:巨噬细胞在一定的条件下可发生炎性程序性细胞死亡,称为焦亡。其中,经典的焦亡途径是依赖NOD样受体蛋白3(NOD-like receptor 3,NLRP3)和半胱氨酸蛋白水解酶1(caspase-1),并释放大量以促炎症因子IL-1β与白细胞介素18(interleukin-18,IL-18)为标志的细胞死亡因子[5]。研究[5-6]显示:巨噬细胞焦亡参与COPD的发生发展过程。近期在脓毒症和急性肺损伤等疾病的研究[7-8]显示:阻断p38 MAPK信号通路可抑制巨噬细胞的焦亡,减轻炎症因子的释放,从而改善疾病的进展。然而,在COPD发生发展过程中,p38 MAPK途径在巨噬细胞焦亡和肺部炎症中的作用尚不清楚。本研究在小鼠体内外采用p38 MAPK抑制剂SB303580阻断该信号通路,探讨COPD的发病机制,为CODP患者的治疗和预后改善提供潜在的作用靶点。
1 材料与方法
1.1 实验动物、细胞、主要试剂和仪器48只SPF级雄性C57BL/6小鼠,体质量(20±5)g,购自海南医学院实验动物中心,动物使用许可证:SYXK(琼)2019-0017;所有小鼠饲养于22℃、12 h光/暗循环的无特定病原体级标准动物房中,自由摄取食物和饮水;本研究动物方案经本院伦理委员会审核通过,所有动物操作均符合实验动物使用和保护条例。小鼠巨噬细胞RAW264.7购自上海众华生物科技公司。红玫卷烟(焦油:13 mg/支;尼古丁:1.3 mg/支)(广东中烟工业有限责任公司),RPMI-1640培养基和1%青-链霉素双抗混合液(美国Hyclone公司),胎牛血清(fetal bovine serum,FBS)(美国Gibco公司),脂多糖(lipopolysaccharide, LPS) 和 乙 酰 甲 胆 碱(methacholine,Mch)(美国Sigma公司),MAPK p38抑制剂SB203580(简称SB)(美国Selleck化学公司),小鼠TNF-α、IL-1β、IL-18、白细胞介素6(interleukin-6,IL-6)ELISA试剂盒(美国Abcam公司),兔抗NLRP3和大鼠抗裂解型半胱氨酸蛋白水解酶1(cleaved caspase-1)(美国Santa Cruz公司),Cy3标记的山羊抗兔IgG和FITC标记的山羊抗大鼠IgG(武汉博士德生物工程有限公司),大鼠抗半胱氨酸蛋白水解酶1前体(procaspase-1)、cleaved caspase-1、裂解型半胱氨酸蛋白水解酶3(cleaved caspase-3)、凋亡相关斑点样蛋 白 (apoptosis-associated speck-like protein,ASC)和Toll样 受 体4(Toll-like receptor 4,TLR4)(美国Bioworld公司),兔抗细胞核因子κB(nuclear factor-κB,NF-κB)p65、p-p38和p-38(美国Cell Signaling Technology公司),抗兔或大鼠IgG、细胞裂解液、蛋白酶抑制剂和二喹啉甲酸(bicinchoninic acid,BCA)蛋白定量试剂盒(江苏碧云天生物技术有限公司),AnnexinⅤ-FITC/PI凋亡试剂盒(美国BD Bioscience公司),FLICA 660-YVAD-FMK caspase-1试剂(美国ImmunoChemistry Technologies公司)。电泳槽、电泳仪和化学发光荧光成像系统(美国Bio-Bad公司),FACSC Calibar流式细胞仪(美国BD Bioscience公司)。
1.2 COPD模型制备和动物分组48只小鼠适应性饲养1周后,随机分为对照组、SB、COPD组和COPD+SB组,每组12只。对照组:正常饲养,暴露于室内空气中;SB组:对正常饲养的暴露于室内空气的小鼠,按20 mg·kg-1在第4~6天给予小鼠腹腔注射溶于DMSO的SB;COPD组:参考文献[9-10]方法,分别于上午09:00~11:00和下午14:00~16:00将小鼠全身暴露于特制烟雾箱中(50 cm×30 cm×30 cm),每小时给予8支烟,间隔1 h予以换气20 min,连续7 d,并于第4天时在麻醉状态下经鼻给予溶于生理盐水的0.2 g·L-1LPS 50μL;COPD+SB组:造模操作同COPD组,但在暴露的第4~6天时按20 mg·kg-1给予小鼠腹腔注射溶于DMSO的SB。此外,对照组和COPD组小鼠在第4~6天给予等体积的DMSO进行腹腔注射。
1.3 各组小鼠气道高反应性(airway hyperresponsiveness,AHR)检测参考文献[11-12]方法以吸入Mch后气道阻力的变化来评价小鼠肺功能改变,在烟雾暴露实验结束24 h后,采用10%水合氯醛麻醉小鼠,切开气管进行插管,首先检测肺阻力基线值(相当于给予5 g·L-1Mch)3 min,然后给予不同浓度(1.5、2.5和5.0 g·L-1)Mch 10μL刺激气道,再次记录肺阻力值。分析各组小鼠不同刺激浓度的平均值,计算并绘制小鼠气道反应曲线,观察各组小鼠的AHR。
1.4 支气管肺泡灌洗液(bronchoalveolar lavage fluid,BALF)的收集和细胞分类计数脱颈处死各组小鼠,分离并暴露支气管,剖开胸腔,结扎右主支气管并摘取右肺后,采用缝合线将注射器针头结扎固定于左主支气管上,采用0.5 mL、4℃预冷的无菌生理盐水灌洗左主支气管和肺泡,反复抽吸3次后,抽出液体,800 g离心5 min,收集上清,-80℃保存用于细胞因子检测。取离心后的细胞沉淀,重悬于200μL PBS溶液中,采用细胞计数板在显微镜下计数细胞总数。随后取约200个细胞采用瑞氏-吉姆萨试剂染色,计数中性粒细胞、巨噬细胞和淋巴细胞数量。
1.5 ELISA法检测BALF上清液中细胞因子水平取“1.4”步骤中收集的BALF上清液,参考相应ELISA试剂盒使用说明方法检测各组小鼠BALF中TNF-α、IL-6、IL-1β和IL-18水平。
1.6 各组小鼠肺组织病理形态观察取各组小鼠右肺组织,采用4%多聚甲醛溶液进行固定24 h后,梯度酒精脱水,石蜡包埋后,切片至4μm厚度,脱蜡,苏木精-伊红(hematoxylin and eosin,HE)染色,于显微镜下观察拍照。
1.7 细胞培养和分组RAW264.7细胞培养于含10%FBS和1%青-链霉素双抗的RPMI-1640培养基中,并置于37℃、5%CO2细胞培养箱中培养。参考文献[11]方法将3支去过滤嘴的香烟烟雾泵入30 mL PBS溶液中以制备烟草提取物(cigarette smoke extract,CSE),待烟雾完全溶解后采用0.22μmol·L-1过滤器进行过滤,在酶标仪320 nm处检测吸光度(A)值并进行标准化后,采 用1 mmol·L-1氢氧化钠调整pH值为7.2~7.4,制备成100%CSE溶液后进行分装,-80℃保存备用。按照实验目的将RAW264.7细胞分为对照组、SB组、CSE组和SB+CSE组。对照组,即正常培养的RAW264.7细胞;SB组:单独采用10 mmol·L-1SB进行处理的RAW264.7细胞;CSE组:100%CSE单独刺激的RAW264.7细胞;SB+CSE组:采 用10 mmol·L-1SB和100%CSE联合处理的RAW264.7细胞。上述各组细胞置于37℃、5%CO2细胞培养箱中培养24 h后进行相关后续实验。
1.8 免疫荧光法检测各组巨噬细胞中NLRP3和cleaved caspase-1表达收集生长状态良好的RAW264.7细胞,常规消化后,调整细胞密度至1×104mL-1,接种至24孔板中,培养12 h待细胞贴壁后按照“1.7”步骤中进行分组处理24 h后,4%多聚甲醛固定细胞,0.5%Triton膜透化处理,1%牛血清白蛋白抗体封闭,随即加入兔抗NLRP3(1∶1 000)与大鼠抗cleaved caspase-1(1∶1 000)一抗在4℃下孵育过夜,次日加入FITC与Cy3标记山羊抗兔与抗大鼠的IgG(1∶1 000),室温下孵育2 h,最后采用DAPI试剂进行细胞核染色10 min,荧光显微镜下观察并拍照。其中细胞中NLRP3阳性表达为绿色荧光信号,cleaved caspase-1阳性表达为红色,细胞核为蓝色。实验单独重复3次。
1.9 流式细胞术检测各组巨噬细胞焦亡率和早期凋亡率收集各组巨噬细胞,取约1×105mL-1细胞重悬于100μL PBS溶液中,依次加入各5μL AnnexinⅤ-FITC和PI试剂,室温下避光孵育20 min,随后采用PBS溶液洗涤细胞后,4%多聚甲醛固定10 min。同时另取约1×105mL-1细胞重悬于150μL PBS溶液中,分别加入AnnexinⅤ-FITC和PI试剂各5μL及FLICA 660-YVAD-FMK caspase-1试剂5μL,室温下同样避光孵育20 min,4%多聚甲醛固定,上机分析组巨噬细胞焦亡率和细胞早期凋亡率。其中焦亡细胞主要特征为caspase-1+PI+,而早期凋亡的细胞为AnnexinⅤ-FITC+PI-。细胞焦亡率=焦亡细胞数/总细胞数×100%,细胞早期凋亡率=早期凋亡细胞数/总细胞数×100%。实验单独重复3次。
1.10 Western blotting法检测各组小鼠肺组织和巨噬细胞中焦亡相关蛋白及NF-κB信号通路相关蛋白表达水平收集待测组织和细胞,采用细胞裂解液和蛋白酶抑制剂提取细胞总蛋白。BCA法检测细胞和组织中的蛋白浓度,加热变性后,取约30μg蛋白样品于10%SDS-PAGE凝胶进行电泳,将分离的蛋白条带采用湿转法转移至PVDF膜上,采用5%脱脂奶粉在室温下进行抗体封闭1 h后,加入NLRP3(1∶500)、pro-caspase-1(1∶1 000)、cleaved caspase-1(1∶1 000)、cleaved caspase-3(1∶1 000)、ASC(1∶500)、NF-κB p65(1∶800)、TLR4(1∶800)、p-38(1∶1 000)和p-p38(1∶1 000)4℃孵育过夜,次日,室温下加入HRP标记的二抗(1∶2 000)孵育1 h,最后滴加ECL发光液曝光拍照,采用Image J图像分析软件进行灰度值分析,计算目的蛋白表达水平。目的蛋白表达水平=目的蛋白条带灰度值/内参蛋白条带灰度值。
1.11 统计学分析采用GraphPad Prism 5.0统计软件进行统计学分析。各组小鼠BALF中各类细胞计数,TNF-α、IL-6、IL-1β和IL-18水平,各组小鼠肺组织中NLRP3/caspase-1和NF-κB信号通路相关蛋白表达水平及各组RAW264.7细胞早期凋亡率和焦亡率均符合正态分布,以±s表示,多组间样本均数比较采用单因素方差分析(One-way ANOVA),组间两两比较采用LSDt检验。以P<0.05为差异有统计学意义。
2 结 果
2.1 各组小鼠肺功能以吸入Mch后气道阻力的变化来评价各组小鼠肺功能改变的结果显示:Mch以剂量依赖性增加各组小鼠的气道阻力,与对照组比较,SB组小鼠气道阻力无明显变化(P>0.05),COPD组小鼠气道阻力明显增加(P<0.05),且以5 g·L-1Mch作用最为明显;与COPD组比较,COPD+SB组小鼠的气道阻力降低(P<0.05)。见图1。

图1 各组小鼠气道阻力Fig.1 Airway resistance of mice in various groups
2.2 各组小鼠肺组织病理形态表现HE染色观察结果显示:对照组和SB组小鼠支气管黏膜上皮及肺泡结构完整,肺泡轮廓清晰,无明显病理性损伤改变。COPD组小鼠肺组织支气管气道黏膜层和肺泡间隔明显增宽,气道上皮细胞受损脱落,出现部分相邻肺泡融合至肺大泡等典型的COPD病理学损伤性改变。与COPD组比较,COPD+SB组小鼠肺组织损伤明显减轻,表现为支气管黏膜上皮及肺泡结构破坏减轻,肺大泡形成明显减少。见图2。
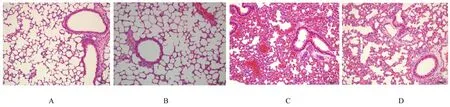
图2 HE染色观察各组小鼠肺组织病理形态表现(Bar=50μm)Fig.2 Pathomorphology of lung tissue of mice in various groups observed by HE staining(Bar=50μm)
2.3 各组小鼠BALF中炎症细胞数量和炎症因子水平BALF中细胞分类计数结果显示:与对照组比较,SB组小鼠BALF中白细胞总数、中性粒细胞数、巨噬细胞和淋巴细胞数量差异无统计学意义(P>0.05),而COPD组和COPD+SB组上述指标均明显升高(P<0.05或P<0.01);与COPD组比较,COPD+SB组小鼠BALF中白细胞总数、中性粒细胞、巨噬细胞和淋巴细胞数量均明显降低(P<0.05)。ELISA法检测结果显示:与对照组比较,SB组小鼠BALF中炎症因子TNF-α、IL-6、IL-1β和IL-18水平均无明显变化(P>0.05),而COPD组小鼠上述炎症因子水平升高(P<0.05或P<0.01);与COPD组比较,COPD+SB组小鼠BALF中TNF-α、IL-6、IL-1β和IL-18水平均降低(P<0.05)。见表1和2。
表1 各组小鼠BALF中炎症细胞数T ab.1 Number of inflammatory cells in BALF of mice in various groups (n=6,±s)

表1 各组小鼠BALF中炎症细胞数T ab.1 Number of inflammatory cells in BALF of mice in various groups (n=6,±s)
*P<0.05 compared with control group;△P<0.05 compared with COPD group.
表2 各组小鼠BALF中炎症因子水平Tab.2 Levels of inflammatory factors in BALF of mice in various groups [n=6,±s,ρB/(ng·L-1)]

表2 各组小鼠BALF中炎症因子水平Tab.2 Levels of inflammatory factors in BALF of mice in various groups [n=6,±s,ρB/(ng·L-1)]
*P<0.05,**P<0.01 compared with control group;△P<0.05,△△P<0.01 compared with COPD group.
2.4 各组巨噬细胞中NLRP3和cleaved caspase-1的表达与对照组比较,SB组巨噬细胞中NLRP3和cleaved caspase-1的荧光表达强度无明显差异,而CSE组巨噬细胞中NLRP3和caspase-1的荧光表达强度均明显升高;与CSE组比较,CSE+SB组巨噬细胞中NLRP3和caspase-1的荧光表达强度均明显降低,提示SB能抑制CSE诱导的巨噬细胞中NLRP3和cleaved caspase-1表达。见图3。
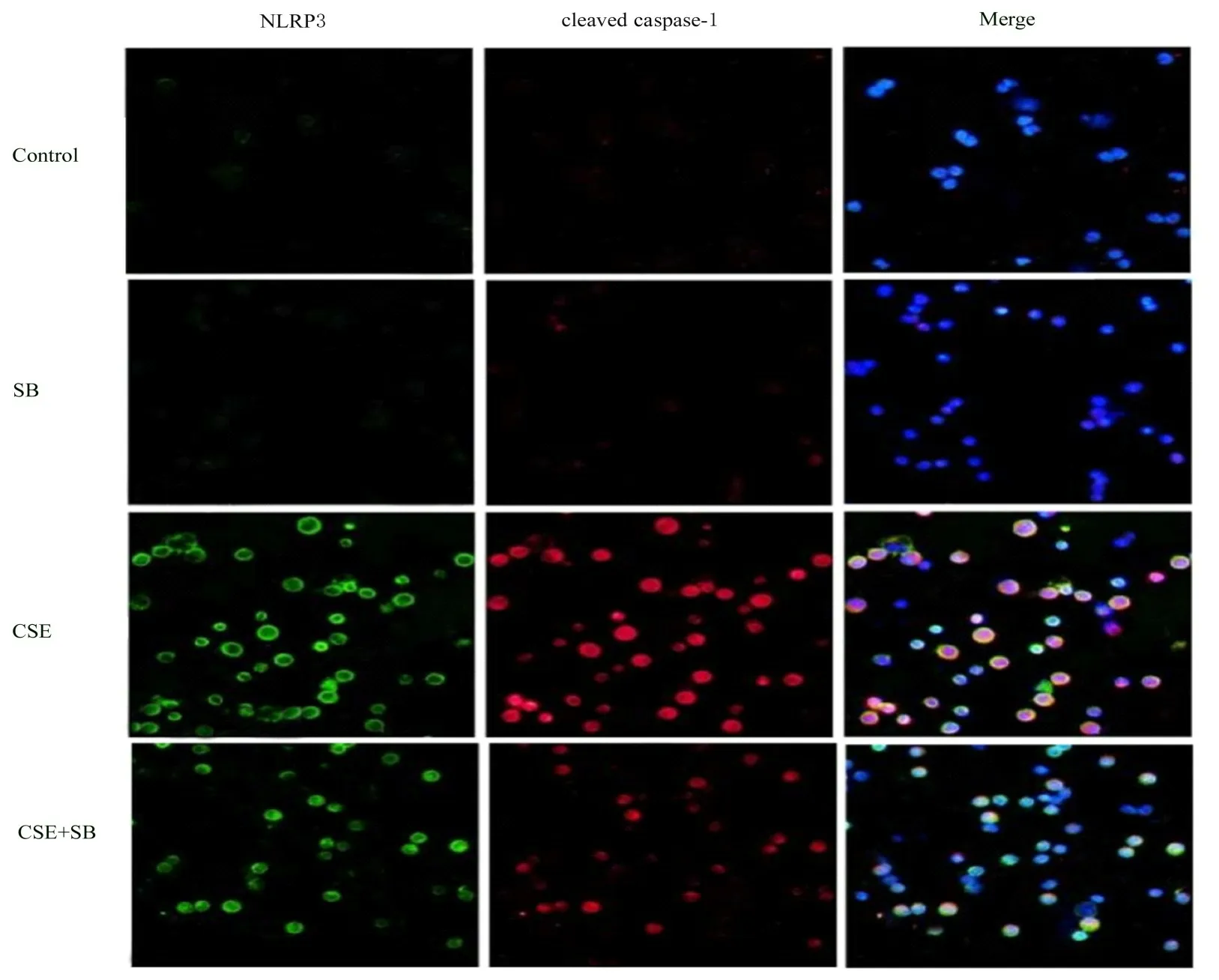
图3 各组巨噬细胞中NLRP3和cleaved caspase-1的表达(免疫荧光,×400)Fig.3 Expressions of NLRP3 and cleaved caspase-1 in macrophages in various groups(Immunofluorescence,×400)
2.5 各组巨噬细胞焦亡率和早期凋亡率流式细胞术检测结果显示:与对照组比较,SB组细胞焦亡率和早期凋亡率差异无统计学意义(P>0.05),但CSE组细胞焦亡率和早期凋亡率均升高(P<0.05);与CSE组比较,CSE+SB组细胞焦亡率降低(P<0.05),但早期凋亡率升高(P<0.05),提示SB能抑制CSE诱导的巨噬细胞中焦亡的发 生,并促进细胞发生凋亡。见图4~7。

图4 流式细胞术检测各组巨噬细胞焦亡率Fig.4 Pyrototic rates of macrophages in various groups detected by flow cytometry
2.6 各组小鼠肺组织中NLRP3/caspase-1和NF-κB信号通路相关蛋白表达水平Western blotting法检测结果显示:与对照组比较,SB组小鼠肺组织中p-p-38蛋白表达水平降低(P<0.05),小鼠肺组织中NLRP3、caspase-1、ASC、NF-κB p65和TLR4蛋白表达水平差异均无统计学意义(P>0.05);COPD组 小 鼠 肺 组 织 中p-p-38、NLRP3、caspase-1、ASC、NF-κB p65和TLR4蛋白表达水平均升高(P<0.05);与COPD组比较,COPD+SB组小鼠肺组织中p-p-38、NLRP3、caspase-1、ASC、NF-κB p65和TLR4蛋白表达水平均降低(P<0.05)。见图8。
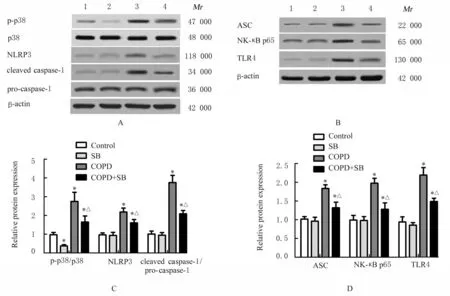
图8 各组小鼠肺组织中NLRP3/caspase-1和NF-κB信号通路相关蛋白表达电泳图(A,B)和直条图(C,D)Fig.8 Electrophoregram(A,B)and histogram(C,D)of expressions of NLRP3/caspase-1 and NF-κB signaling pathway related proteins in lung tissue of mice in various groups
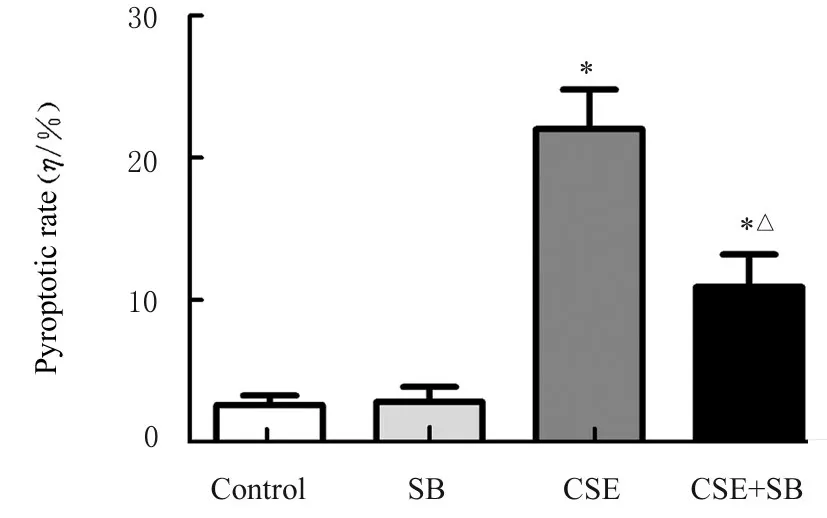
图5 各组巨噬细胞焦亡率Fig.5 Pyroptotic rates of macrophages in various groups
2.7 各组巨噬细胞中MAPK和NF-κB信号通路相关蛋白表达水平Western blotting法检测结果显示:与对照组比较,SB组巨噬细胞中p-p-38/p-38比值明显降低(P<0.05),但NF-κB信号通路中NF-κB p65、TLR4和cleaved caspase-3蛋白表达水平差异均无统计学意义(P>0.05),而CSE组和CSE+SB组巨噬细胞中p-p-38/p-38比值、NF-κB p65、TLR4和cleaved caspase-3蛋白表达水平均升高(P<0.05或P<0.01);与CSE组比较,CSE+SB组巨噬细胞中p-p-38/p-38比值、NF-κB p65和TLR4表达水平均降低(P<0.05),但cleaved caspas-3蛋白表达水平升高(P<0.05)。见图9。

图6 流式细胞术检测各组巨噬细胞早期凋亡率Fig.6 Early apoptotic rates of macrophages in various groups detected by flow cytometry

图7 各组巨噬细胞早期凋亡率Fig.7 Early apoptotic rates of macrophages in various groups

图9 各组小鼠肺组织中MAPK和NF-κB信号通路蛋白表达电泳图(A)和直条图(B)Fig.9 Electrophoregram(A)and histogram(B)of expressions of MAPK and NF-κB signaling pathway related proteins in lung tissue of mice in various groups
3 讨 论
流行病学调查[13]显示:在我国40岁以上人群中,COPD的患病率约为8.2%,并已成为我国农村居民死亡原因的前三位,造成严重的家庭与社会负担。本研究体内实验结果表明:烟雾联合LPS暴露能够显著影响COPD小鼠模型肺部炎症细胞的浸润及炎症因子IL-1β和IL-18的表达,而p38 MAPK抑制剂SB可能通过抑制巨噬细胞焦亡改善COPD肺损伤及炎症反应。
有研究[5]证实:吸烟作为COPD的主要致病因素,能够激活炎性小体,启动细胞焦亡。在动物模型[14]中发现:与正常C57BL/6小鼠比较,在同等剂量和时间的烟雾暴露下,NLRP3基因敲除可明显改善小鼠肺功能的损伤,同时BALF中炎症因子TNF-α、IL-6和IL-1β水平也明显降低。临床研究[15-16]显示:COPD患者血清和肺组织中IL-1β和IL-18水平升高与其全身炎症水平及炎症急性加重具有相关性。本研究体内实验结果同样表明:烟雾联合LPS暴露能够增加肺部炎症细胞的浸润和IL-1β及IL-18的表达;提示细胞焦亡与CODP的发生发展密切相关。而焦亡是由微生物感染或非感染性刺激(如化疗和炎症等)引起的宿主细胞程序性死亡的重要途径之一,其普遍存在于单核细胞、巨噬细胞和树突状细胞中[4,17]。研究[17]显示:细胞焦亡具有不同于细胞坏死或凋亡的独特形态和生化特征,其主要表现为焦亡是caspase-1依赖的,核酸酶介导的DNA裂解,而不具有与凋亡相关的寡合体碎片化模式特征。同时,焦亡伴随着细胞中钾离子的外流和胞内渗透压的降低,并随之发生细胞的肿胀、质膜孔的形成和促炎性胞内容物的释放,而上述胞膜的形态改变使细胞对PI的染色呈阳性反应[18]。进一步的分子机制研究[17-19]显示:在发生焦亡前,细胞中形成的NLRP3炎性小体蛋白复合物与ASC发生寡聚化反应,随即招募大量的pro-caspase-1并诱导其自身发生蛋白水解从而活化为cleaved caspase-1。
为进一步明确细胞焦亡在COPD发生发展中的致病机制,本研究体外培养小鼠巨噬细胞系RAW264.7细胞的实验结果表明:CSE能够刺激巨噬细胞中NLRP3和caspase-1的表达,且明显促进了caspase-1介导的细胞焦亡的发生。而巨噬细胞焦亡的研究最初集中在由病原微生物引起的感染相关疾病中,但近年来研究[20]显示:巨噬细胞焦亡同样参与其他器官或系统的疾病,如在肝组织中的巨噬细胞和Kupffer细胞的焦亡可诱导慢性肝炎与肝纤维化的发生[21];而滑膜巨噬细胞的焦亡参与膝关节炎的发生发展。在呼吸系统中,肺泡巨噬细胞中NLRP3炎症小体的激活导致caspase-1和IL-1β的活化,进一步加重了呼吸窘迫综合征患者机械通气时的肺损伤症状[22]。而caspase-1基因缺失或抑制p38 MAPK信号通路可通过抑制巨噬细胞的焦亡而减轻LPS诱导的小鼠急性肺损伤[8,23]。
p38 MAPK和NF-κB信号通路被认为是炎症反应进程中的主要途径,其能够被各种因素所激活,并最终产生细胞因子、趋化因子和蛋白水解酶,从而加剧炎症反应[24]。既往研究[25]显示:吸烟能够通过增强p38 MAPK和NF-κB信号通路参与COPD疾病中的肺部炎症。研究[9,26]显示:p38 MAPK抑制剂SB能够在体内外降低香烟烟雾对COPD模型诱发肺气肿和炎症因子释放的敏感性,提示抑制p38 MAPK途径可能是COPD治疗的潜在靶标。同时,最近的一项研究[8]显示:干扰p38 MAPK信号通路能够通过改善NF-κB信号通路诱导巨噬细胞焦亡,从而缓解急性肺损伤中的炎症反应。而脓毒败血症诱导的脑损伤小鼠模型研究[7]中发现:人克拉拉细胞蛋白16(clara cell protein 16,CC16)能够通过降低p38 MAPK的磷酸化水平抑制大脑皮质的焦亡,从而发挥保护作用。在本研究中的体内实验同样发现:SB能够改善NLRP3/caspas-1途径介导的巨噬细胞焦亡,明显降低肺组织与巨噬细胞中NLRP3、caspase-1和ASC蛋白的表达,下调细胞中PI的阳性表达率,并抑制NF-κB信号途径的活化。同时,本课题组采用体外细胞实验进一步提示:SB这一作用可能是通过抑制CSE诱导的细胞焦亡,并促进巨噬细胞凋亡的发生,从而减少炎症因子的释放。凋亡能够有效防止细胞中促炎因子、蛋白酶和活性氧等因子的泄露,利于肺组织中炎症的局限和自我稳态的维持[27-28]。已有研究[29]证实:阻断p38 MAPK信号通路可促进有害因素诱导的巨噬细胞凋亡。
综上所述,SB可能改善COPD肺损伤和炎症反应,其机制可能与抑制巨噬细胞焦亡有关。本研究结果进一步阐释了COPD进展的分子机制,而调控p38 MAPK信号通路有望成为改善COPD患者预后的新型靶点。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国医学物理学杂志(2022年9期)2022-10-09
中国现代医生(2022年19期)2022-08-25
军事文摘(2022年8期)2022-05-25
学苑创造·A版(2020年9期)2020-10-13
祝您健康(2018年10期)2018-10-11
金山(2009年11期)2009-01-05
销售与管理(2006年9期)2006-09-17
