
非布司他荧光药物分子的合成及体外活性测试
2022-06-17 08:22汪绪龙姜开元杨文宁
合成化学 2022年6期
汪绪龙, 姜开元, 刘 洋, 杨文宁, 隋 强
(1.上海工程技术大学 化学化工学院,上海 201620; 2.中国医药工业研究总院,上海 201203; 3.北京中医药大学,北京 102488)
非布司他(Febuxostat,12)为黄嘌呤氧化酶(XOD)的选择性抑制剂[1-6],临床用于降低痛风患者的血尿酸。与别嘌醇相比,非布司他对XOD的抑制作用和选择性更好[1],但仍存在不良反应[2]。因此其与靶蛋白在体内的相互作用机制值得深入研究,为后期开发作用效果更强,副作用更小的药物分子奠定基础。
Chart 1
近年来,小分子荧光探针在光学、生物学、分析领域有着广泛的应用。在众多的荧光染料中,硼氟二吡咯化合物(“BODIPY”)因具有荧光量子产率高、高消光系数、光稳定性好、结构易于修饰等优点而被广泛关注。例如,荧光试剂BODIPY-FL[7-9]可用于载药型的荧光探针。5-羟色胺受体“格拉司琼”与BODIPY-FL通过吲哚基团相连后,可转化为一种5-羟色胺探针13,并在细胞成功显影[10]。三萜羧酸类化合物与BODIPY-FL通过乙二胺酰胺缩合连接形成探针14;14对人乳腺腺癌细胞具有细胞毒性,但对其他细胞系无细胞毒性[11]。Bruton’s酪氨酸激酶抑制剂(依鲁替尼)与BODIPY-FL通过酰胺缩合反应转化为依鲁替尼荧光探针15,该探针具有较高的选择性[12]。
在大多数荧光药物分子中,荧光染料和药物分子可通过酰胺键和酯键两种方式结合。非布司他分子结构中噻唑环上的羧基为与酶结合的头部,且与酶结合最为紧密,不宜与BODIPY-FL结合。相对而言,尾部的异丁基与酶结合较为疏松,可提供疏水环境[13-14]。非布司他的活性代谢物(中间体10),与非布司他具有相似的活性[15]。因此,设计将10与BODIPY-FL以酯键结合得到目标化合物(化合物11)。
本文以2-(3-氰基-4-羟基苯基)-4-甲基-1,3-噻唑-5-羧酸乙酯为原料,经水解得到中间体10;以二甲基膦乙酸苄酯为原料,经Witting反应、氢化还原双键、环合及钯/碳催化氢化脱苄基等4步反应,得到中间体BODIPY-FL,最后将BODIPY-FL的羧基和中间体10的羟基酯化缩合得到非布司他荧光分子(Scheme 1),其结构经1H NMR, MS(ESI)表征。采用酶促反应法测定了荧光分子对酶活性的抑制效果[16]。
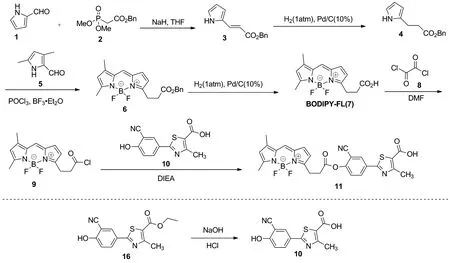
Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker Avance NEO 600 MHz型核磁共振仪(TMS 为内标);Alliance 2695/2486/Q-TOF micro型质谱仪;TU-1810型紫外-可见分光光度计;Starter 2100型pH计。
黄嘌呤,阿拉丁试剂(上海)有限公司;黄嘌呤氧化酶,西格玛奥德里奇(上海)贸易有限公司;其余所用试剂均为分析纯。
1.2 合成
(1)化合物3的合成
将二甲基膦乙酸苄酯287.0 g(0.337 mol)溶于500 mL THF中,冰浴降温至0 ℃,分批加入NaH 4.49 g(0.187 mol),加毕,撤除冰浴,升温至室温,搅拌5 min;冷却至0 ℃,滴加2-吡咯甲醛116.8 g(0.177 mol)的THF(80 mL)溶液,滴毕,撤除冰浴,搅拌下于室温反应90 min。冷却至0 ℃,加入5%柠檬酸溶液200 mL和水500 mL,用乙酸乙酯(3×400 mL)萃取水层,合并有机相,依次用水300 mL和盐水300 mL洗涤,用MgSO4干燥,于37 ℃减压除去溶剂,粗产物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=9/1,V/V)纯化得白色固体331.7 g,收率79%, m.p.25~31 ℃;1H NMR(600 MHz, DMSO-d6)δ: 11.53(s, 1H), 7.52(d,J=15.6 Hz, 1H), 7.40~7.32(m, 5H), 7.04(m, 1H), 6.59(m, 1H), 6.28(d,J=15.6 Hz, 1H), 6.17~6.16(m, 1H), 5.18(s, 2H); MS(ESI)m/z: Calcd for C14H13NO2{[M+Na]+}250.09, found 250.11。
(2)化合物4的合成
将化合物3 10.0 g(0.0440 mol)溶于80 mL 乙酸乙酯中,加入10%Pd/C 0.600 g,常压氢化反应24 h。过滤除去Pd/C,于37 ℃减压下除去溶剂,粗产物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=95/5,V/V)纯化得无色油状物43.80 g,收率38%;1H NMR(600 MHz, DMSO-d6)δ: 10.47(s, 1H), 7.37~7.23(m, 5H), 6.52(m, 1H), 5.80(m, 1H), 5.68(m, 1H), 5.03(s, 2H), 2.76(m, 2H), 2.59(m, 2H); MS(ESI)m/z: Calcd for C14H15NO2{[M+Na]+}252.11, found 252.26。
(3)化合物6的合成
将化合物4 3.00 g(0.0131 mol)和3,5-二甲基-1H-吡咯-2-甲醛51.68 g(0.0136 mol)溶于80 mL PhCF3中,冰浴降温至0 ℃,缓慢滴加POCl34.50 g(0.0293 mol),滴毕,撤除冰浴,升温至室温,搅拌3 h;冷却至0 ℃,缓慢滴加BF3·Et2O 10.9 g(0.0768 mol)和DIEA(N,N-二异丙基乙胺)11.3 g(0.0874 mol),滴毕,撤除冰浴,搅拌下于室温反应12 h。冷却至0 ℃,加入饱和NaHCO3溶液30 mL,用乙酸乙酯(2×100 mL)萃取水层,用盐水100 mL 洗涤,用Na2SO4干燥,于37 ℃减压除去溶剂,粗产物经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=19/1,V/V)纯化得红色固体62.20 g,收率44%, m.p.115~120 ℃;1H NMR(600 MHz, DMSO-d6)δ: 7.63(s, 1H), 7.34~7.25(m, 5H), 7.03(d,J=6.0 Hz, 1H), 6.32(d,J=6.0 Hz, 1H), 6.26(s, 1H), 5.08(s, 2H), 3.10(t,J=11.4 Hz, 2H), 2.76(dd,J=12.6 Hz, 10.8 Hz, 2H), 2.43(s, 3H), 2.21(s, 3H); MS(ESI)m/z: Calcd for C21H21BF2N2O2{[M+Na]+}405.17, found 404.98。
(4)化合物7的合成
将化合物61.67 g(4.37 mmol)溶于100 mL MeOH中,加入10%Pd/C 0.200 g常压氢化反应3 h。过滤除去Pd/C,于35 ℃减压下除去溶剂,粗产物经硅胶柱层析(洗脱剂:三氯甲烷/甲醇=19/1,V/V)纯化得黑亮红色固体BODIPY-FL(7)0.800 g,收率63%, m.p.110~113 ℃;1H NMR(600 MHz, DMSO-d6)δ: 12.29(s, 1H), 7.71(s, 1H), 7.10(d,J=4.2 Hz, 1H), 6.36(d,J=4.2 Hz, 1H), 6.31(s, 1H), 3.08(t,J=7.8 Hz, 2H), 2.65(t,J=7.8 Hz, 2H), 2.48(s, 3H), 2.27(s, 3H); MS(ESI)m/z: Calcd for C14H15BF2N2O2{[M+Na]+}315.12, found 315.14。
(5)化合物10的合成
将2-(3-氰基-4-羟基苯基)-4-甲基-1,3-噻唑-5-羧酸乙酯161.00 g(3.46 mmol)溶于20 mL无水乙醇中,加入1 mol/L NaOH 10.4 mL(10.4 mmol)于60 ℃反应2 h。停止加热,降至室温,加入水20 mL,用1mol/L HCl调pH为3.5左右,用乙酸乙酯(3×40 mL)萃取水层,合并有机相,用Na2SO4干燥,于37 ℃减压除去溶剂,得淡黄色固体100.800 g,收率88%, m.p.230~234 ℃;1H NMR(400 MHz, DMSO-d6)δ: 8.17(d,J=2.4 Hz, 1H), 8.08(dd,J=2.4 Hz, 3.6 Hz, 1H), 7.13(d,J=8.8 Hz, 1H), 2.64(s, 3H); MS(ESI)m/z: Calcd for C12H8N2O3S{[M+H]+}261.03, found 260.99。
(6)化合物11的合成
将BODIPY-FL(7)0.100 g(0.340 mmol)溶于5 mL超干的二氯甲烷中,氮气保护,冰浴降温至0 ℃,缓慢滴加草酰氯80.214 g(1.69 mmol),滴加1滴DMF,加毕,撤除冰浴,升温至室温,搅拌30 min,于32 ℃减压除去溶剂,得到中间体9。将中间体9溶于20 mL超干二氯甲烷中,冰浴降温至0 ℃,滴加化合物10 88.0 mg(0.340 mmol)的DMF(2 mL)和DIEA 0.194 g(1.50 mmol)溶液,滴毕,撤除冰浴,搅拌下于室温反应2 h。冷却至0 ℃,缓慢滴加7%HCl 20 mL,用二氯甲烷(2×40 mL)萃取水层,合并有机相,用Na2SO4干燥,于34 ℃减压除去溶剂,粗产物经硅胶柱层析(洗脱剂:二氯甲烷/甲醇=40/1,V/V)纯化得橙色固体1130.0 mg,收率16%, m.p.161~166 ℃;1H NMR(600 MHz, CDCl3)δ: 8.33(d,J=1.8 Hz, 1H), 8.19(m, 1H), 7.44(d,J=9.0 Hz, 1H), 7.13(s, 1H), 6.90(d,J=3.6 Hz, 1H), 6.38(d,J=4.2 Hz, 1H), 6.15(s, 1H), 3.46(t,J=7.2 Hz, 2H), 3.19(t,J=7.8 Hz, 2H), 2.82(s, 3H), 2.60(s, 3H), 2.28(s, 3H); MS(ESI)m/z: Calcd for C26H21BF2N4O4S{[M+Na]+}557.13, found 557.22。
1.3 体外活性测试
(1)实验步骤
取200 μL黄嘌呤氧化酶(50 nM,PBS稀释)加入2 mL EP管中,分别加入200 μL系列不用浓度非布司他荧光探针,室温预温孵5 min后加入600 μL 底物黄嘌呤(62.5 μmol,PBS稀释)启动反应,反应5 min,于298 nm处测定吸光度值A,一式两份,反应体系中DMSO含量低于0.1%。以只有底物黄嘌呤的缓冲液作为空白组,记录实验数据并计算抑制率。
(2)实验结果
采用Graphpad Prism 8.3.0软件对抑制剂浓度和相应的抑制率进行拟合,应用四参数Logistic模型,求解得到非布司他荧光探针的IC50为8.3 nM。
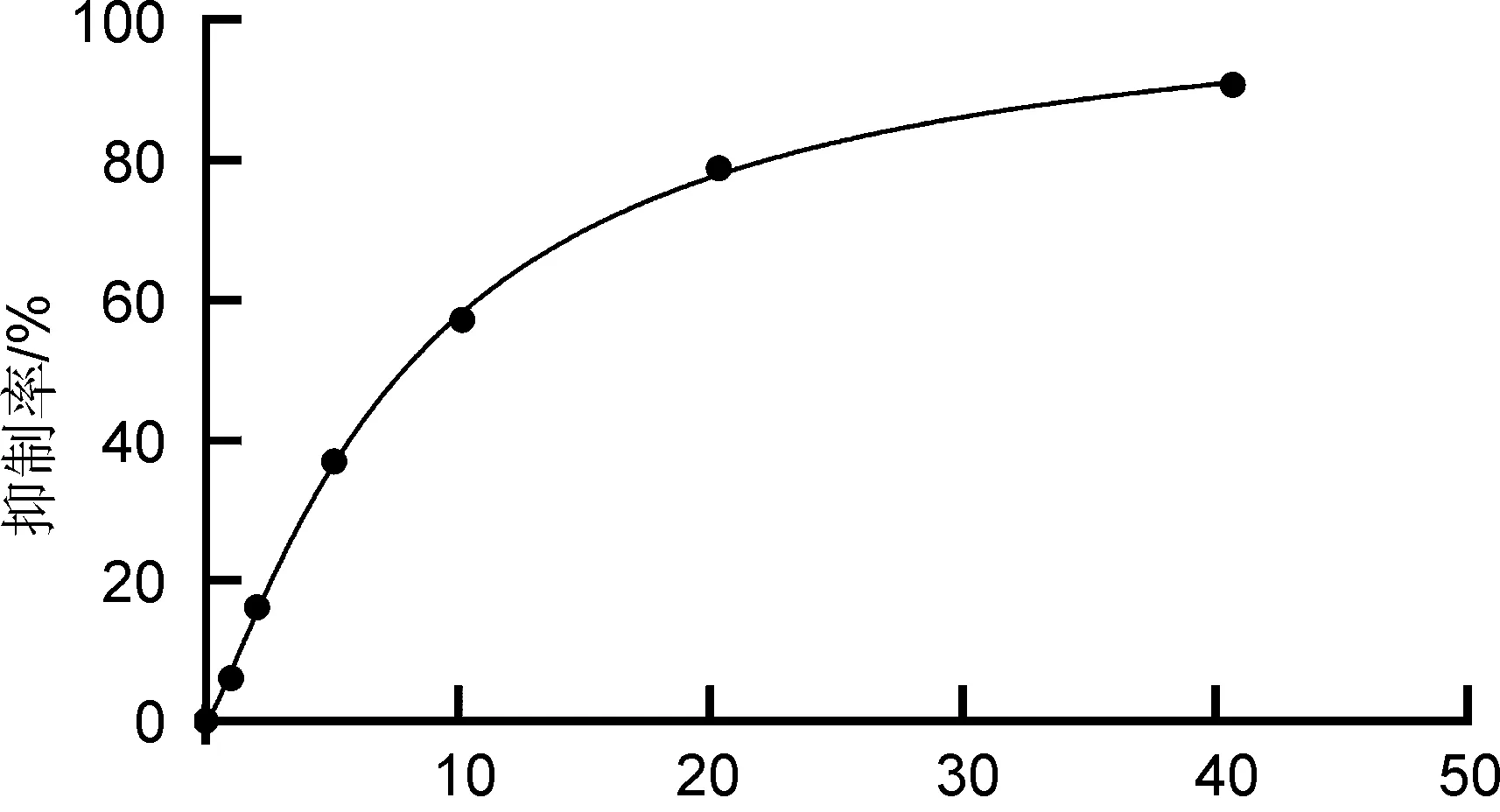
抑制剂浓度/nmol
2 结果与讨论
2.1 合成
化合物3发生氢化反应时,有双键和苄基两个反应位点,为了避免苄基脱除,合成化合物4时在反应体系中加入微量的Ph2S(二苯硫醚)使Pd/C毒化,但是,存在副产物和反应不完全,导致收率低。为了解决这一问题,筛选不同反应条件,如氢化溶剂、反应压力和反应体系有无Ph2S,研究发现当选用乙酸乙酯为反应溶剂、常压和反应体系无Ph2S时,收率有所提高。设计采用氯化亚砜来制备中间体9,但实验过程中发现减压浓缩不易除去多余的氯化亚砜且容易使中间体9降解,因此,改用低沸点的草酰氯来制备中间体9,避免产物的降解,成功得到的目标化合物(化合物11)。相对于其他通过酰胺缩合或者通过其他基团连接得到的荧光分子,本文通过BODIPY-FL与化合物10直接酯化,得到了非布司他荧光药物分子,尽量减少干扰化合物10的生物活性。
2.2 体外活性
根据前期实验结果,在该酶促反应体系条件下,非布司他的IC50值约为2 nM[17],此次测得的非布司他荧光药物分子的IC50值为8.3 nM,说明接入荧光基团后其与黄嘌呤氧化酶的亲和力略有下降,但依然具有很强的亲和力,可以作为探针分子用于后期研究非布司他与XOD在体内靶蛋白处的动态结合与解离过程。
以二甲基膦乙酸苄酯为原料,经Witting反应、氢化还原双键、环合及钯/碳催化氢化脱苄基四步反应,得到BODIPY-FL荧光染料,进一步设计合成了非布司他荧光药物分子,并对其结构进行了表征。测定了该荧光药物分子对XOD的抑制效果,结果表明合成的非布司他荧光药物分子能与XOD紧密结合,亲和力大。这种基于荧光探针构建具有诊断和靶向治疗作用的药物分子,可为后期研究非布司他与靶蛋白在体内的动态结合过程提供参考。
猜你喜欢
世界农药(2022年10期)2022-11-10
河南医学研究(2022年16期)2022-09-01
医学概论(2022年4期)2022-04-24
云南化工(2022年2期)2022-03-18
能源化工(2021年2期)2021-12-30
昆明医科大学学报(2021年8期)2021-08-13
河北医科大学学报(2021年3期)2021-04-06
农药科学与管理(2021年2期)2021-03-16
大众健康(2019年11期)2019-01-19
读与写·下旬刊(2018年6期)2018-07-14
