
家族性扩张型心肌病3例并文献复习
2022-06-08 14:02谭丽陈然
中国现代医生 2022年10期
谭丽 陈然

[摘要] 家族性扩张型心肌病(familial dilated cardiomyopathy,FDCM)是指一个家系中包含先证者中有两个及两个以上确诊扩张型心肌病的患者,或者先证者家系中有小于35岁的一级亲属猝死史。由于FDCM具有遗传多样性、遗传异质性、不完全外显率或年龄相关外显率的特征,导致临床医师经常忽视FDCM的诊断。本文详细介绍了一个家系中Ⅱ-2、Ⅱ-4、Ⅱ-7成员在临床随访过程中先后出现气促症状,完善心脏彩超、冠状动脉造影后,被确诊为FDCM的过程,并对FDCM的诊治进行系统性文献复习,希望广大医师提高对FDCM的认识。
[关键词] 扩张型心肌病;家族性疾病;遗传因素;临床诊断
[中图分类号] R522.1 [文献标识码] B [文章编号] 1673-9701(2022)10-0174-03
[Abstract] The familial dilated cardiomyopathy (FDCM) is defined as a family with two or more proband diagnosed with dilated cardiomyopathy (DCM), or a family with a history of sudden death in first-degree relatives younger than 35 years old. FDCM is characterized by genetic diversity, genetic heterogeneity, incomplete penetrance, or age-related penetrance, leading clinicians to often overlook the diagnosis of FDCM. This paper introduces in detail how members of a family,Ⅱ-2, Ⅱ-4 and Ⅱ-7, developed shortness of breath during clinical follow-up and were diagnosed with FDCM after completing cardiac color doppler ultrasound and coronary angiography, and systematically reviews the diagnosis and treatment of FDCM in the hope that doctors can improve their understanding of FDCM.
[Key words] Dilated cardiomyopathy (DCM); Familial disease; Hereditary factor; Clinical diagnosis
擴张型心肌病(dilated Cardiomyopathy,DCM)指在没有高血压、瓣膜病或冠状动脉疾病等异常负荷条件下,以心室扩大、左心功能降低为特征的一类心肌病。研究调查表明25%~50%的DCM为聚集性发病。在一个家系中包含先证者在内有两个或以上DCM的患者,或者先证者家系中有小于35岁的一级亲属猝死史,可以考虑为FDCM。基因突变与FDCM密切相关,目前已经发现至少60个基因与FDCM有关,包括肌联蛋白基因、细胞核膜基因、细胞骨架蛋白基因等,它们编码心肌细胞肌小节、核膜和细胞骨架中多种蛋白质,保证心肌细胞的正常生理功能[1]。上述基因发生突变造成蛋白合成异常,最终导致FDCM的发生。FDCM具有遗传与临床表现异质性及不完全外显率或年龄相关外显率的特征,使得其早期诊断具有一定的挑战性[2]。本文通过该院收治的3例FDCM患者,并对FDCM进行系统性文献复习,现报道如下。
1 临床资料
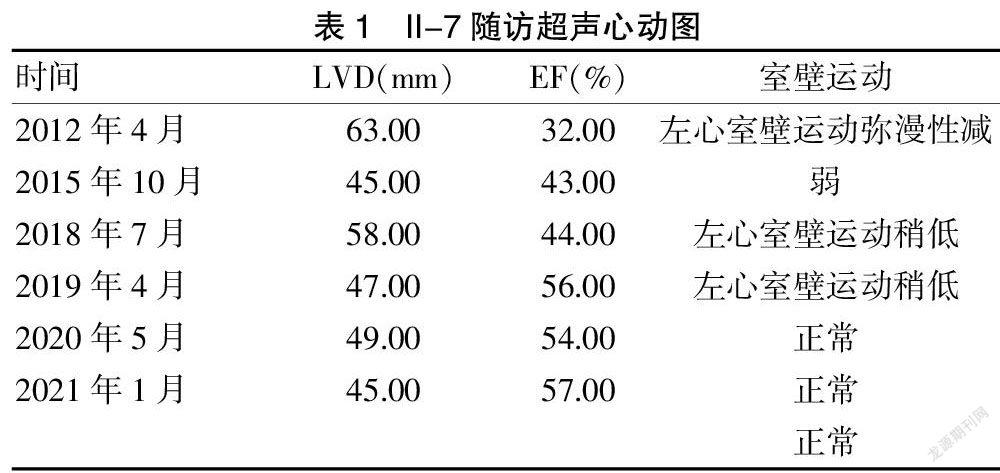
病例1:先证者Ⅱ-7,2012年4月因“活动后气促3个月”就诊,查BNP 716.00 pg/ml,心脏彩超:左心增大[左心室舒张末期内径(LVED)为63.00mm],心功能降低[射血分数(EF)为32.00%)],左心室壁运动弥漫性减弱。心电图:窦性心动过速。冠脉造影正常。诊断为DCM。出院后长期口服呋塞米、螺内酯、美托洛尔缓释片、贝那普利抗心力衰竭治疗,随访9年,症状逐渐好转,心脏彩超如表1。
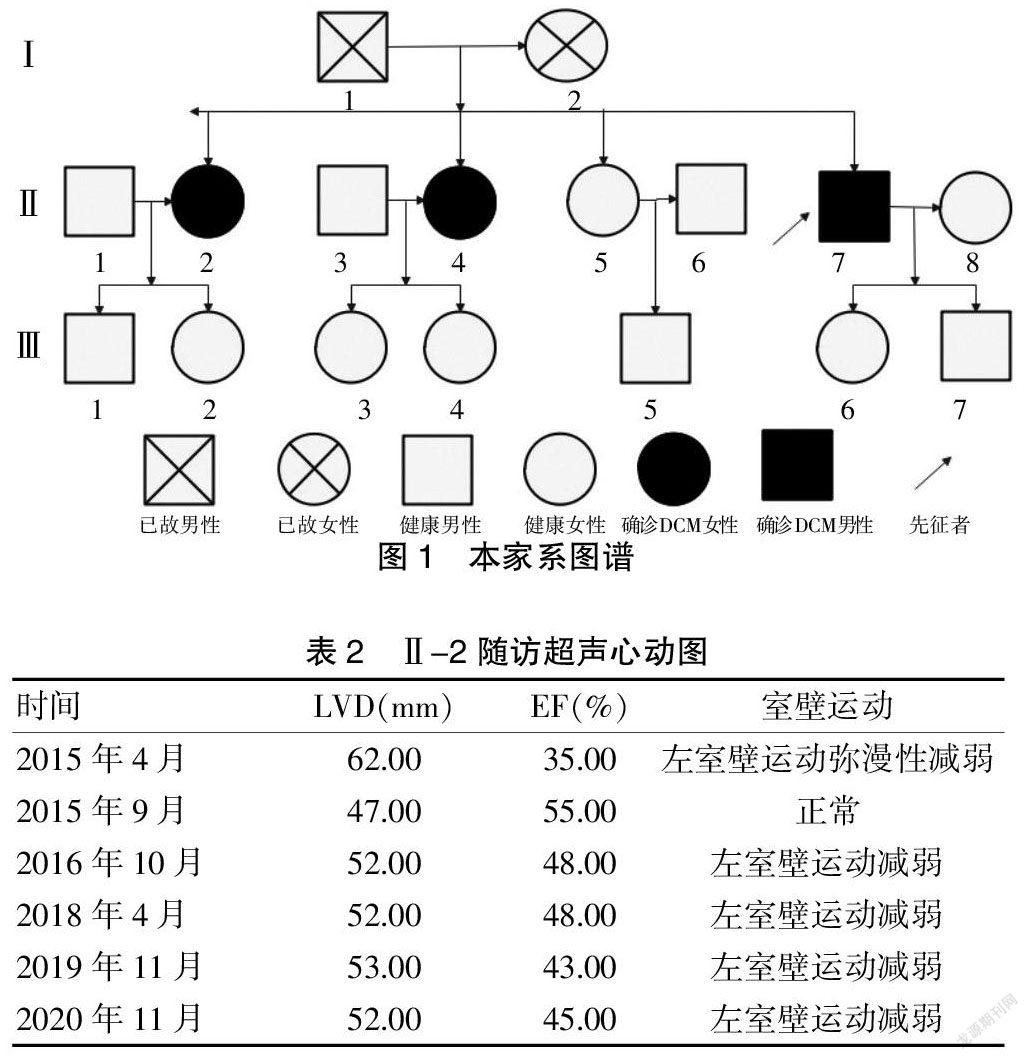
病例2 先证者姐姐Ⅱ-2,2015年4月因“活动后气促1月”就诊,BNP 920.00pg/ml,心脏彩超:左心大(LVED为62.00 mm),左心功能降低(EF为35.00%),左心室壁运动弥漫性减弱。心电图:窦性心动过速。冠脉造影:左前降支狭窄约40.00%。诊断考虑DCM,出院后长期口服呋塞米、螺内酯、美托洛尔缓释片、贝那普利抗心力衰竭治疗,随访6年,气促症状间断加重,心脏彩超如表2。
病例3 先证者姐姐Ⅱ-4,2016为10月体检心电图及心脏彩超均正常。2021年3年因“胸闷气促1周”就诊,BNP 2989.00 pg/ml,心脏彩超:左心增大(LVED为51.00 mm),左心功能减低(LVEF为44.00%),左心室壁运动弥漫性减弱。冠脉造影未见异常。诊断为DCM。出院后长期服用沙库巴曲缬沙坦、美托洛尔缓释片、呋塞米、螺内酯抗心力衰竭治疗,随访2个月,气促症状好转,心脏彩超如表3。
3 讨论
3.1 病例讨论
该先证者为一名中年男性,家族图谱见图1。其母亲51岁不明原因猝死,父亲70岁死于脑出血,先证者及2位姐姐分别于42岁、46岁、55岁出现典型的活动后气促症状;心脏彩超均可见心脏扩大,伴有收缩功能障碍,在排除其他类型心脏病及感染、中毒或环境因素等获得性病因,可以被诊断为FDCM。
3.2 FDCM的特点
FDCM发病年龄通常在20~50岁之间,失代偿性心力衰竭是最常见的临床表现[3]。其遗传方式多样,大多数突变为常染色体显性遗传[4]。FDCM存在高度遗传异质性,家系成员即使携带相同致病基因疾病特点不尽相同,这可能与遗传基因具有外显率不全或年龄相关外显率的特点有关;且不同的基因位点突变也可以导致相同的临床表型[5]。上述证据支持FDCM被环境和遗传因素共同影响的观点。
3.3 FDCM的诊断
研究发现FDCM较散在性患者的预后更差,早期诊断FDCM有助于改善患者预后。但FDCM家族成员中携带致病突变的个体可无临床表现,仅出现心脏结构异常;随着年龄的增长,症状逐渐出现,很多FDCM的首例患者往往被忽视。因此对合并以下特征的散发性DCM患者我们应引起重视:①发病年龄<35岁;②有心源性猝死家族史;③伴发心电传导疾病;④伴有心外表现: 如神经肌肉疾病、畸形、智力残疾[6]。
对于怀疑FDCM的患者,三代家族史以及一级亲属的临床筛查是必要的,基因检测也占据举足轻重的位置。另外,斑点追踪、应变和应变率、MRI以及非编码 RNA 等循环生物学标志物,已成为帮助心脏病专家临床实践的潜在策略。
3.3.1一级亲属筛查 病史和临床影像学检查是FDCM诊断的基石。心电图是最简单的检查,用于评估心脏传导功能。超声心动图是最重要的检查,主要通过左心室舒张末期前后直径和左心室射血分数等指标评估心功能。Verdonschot等[7]研究证明: DCM亲属的LVEF正常队列的心室整体纵向应变(global longitudinal strain,GLS)较正常患者明显降低,并在随访过程中发现异常的GLS与LVEF的降低相关。GLS似乎是一种很有前景的DCM亲属筛查工具。
对于未行基因检测的家系,ESC指南建议筛查应从10bolangx12岁开始,每1~2年进行心脏评估,20岁以后每2~5年进行心脏评估,直到60~65岁。有基因结果的家系,对于携带致病基因变异的个体,ESC指南建议1~3年进行心脏评估;AHA指南则建议3~5年进行心脏评估[8]。非致病基因携带者不需要临床随访。目前指南意见尚未统一,需要进一步研究,有更多的数据支持来帮助临床医师更好地诊断FDCM。
3.3.2 基因检测 基因检测在FDCM患者中具有重要地位,指南建议将基因检测作为家族性病例诊断的一部分。TTN基因变异是FDCM最常见的原因,约占FDCM的30%~35%[9];与 FDCM 相关的第二個最常见的突变是在 LMNA 基因中发现的,大约占10%~15%[10]。近年来随着分子生物学和二代基因测序(next-generation sequencing, NGS)的发展,早期诊断FDCM变为可能[11]。但是由于地域、经济以及医保的限制,有部分人未接受基因检测。且在接受全面基因检测的患者中,有超过一半的患者没有得到分子诊断,除了TTN基因外的大部分基因预期诊断率都没有超过5%[12]],使遗传诊断复杂化。
另外对于检测结果的解读也尤为重要。美国医学遗传学和基因组学将变异体分为五个等级:致病性、可能致病性、未知致病性、可能良性和良性。只有 “致病”和“可能致病”被认为具有临床意义。
3.4治疗
FDCM一般以心力衰竭、心律失常为常见的首发表现,近年来尽管在DCM的心衰管理上取得了一定的进展,但尚无彻底的治疗方法[13],其治疗旨在控制临床症状、改善心功能及生存质量,降低死亡率。
3.4.1药物治疗 药物治疗是治疗心力衰竭的基石,在接受标准药物治疗的家族性和非家族性DCM中,5年生存率无显著差异[14]。2021年ESC指南更新建议:心力衰竭患者无药物禁忌证的情况下,予血管紧张素转换酶抑制剂 (ACEI) 或沙库巴曲缬沙坦(ARNI)联合β受体阻滞剂及盐皮质激素受体拮抗剂 (MRA)标准抗心力衰竭治疗方案。无论是否合并糖尿病,均应加用达格列净或恩格列净降低心血管事件风险[15]。
3.4.2非药物治疗 近年来器械治疗逐渐发展,对FDCM的治疗有很大的帮助。主要包含心力衰竭的心脏再同步化治疗(CRT)、植入型心律转复除颤器(ICD)及左心室辅助装置(LVAD)。心脏移植为最终治疗方法,但难以广泛开展。
本文通过系统性文献复习介绍了FDCM的诊治,希望广大医师提高对FDCM的认识。在接诊DCM患者的过程中,应详细采集家族史。特别是对于发病年龄轻、家族中有不明原因猝死或者发病原因不明的DCM患者,临床医师需提高警惕,可建议患者及其一级亲属进行相应的筛查。
[参考文献]
[1] 李雪银,李广平.家族性扩张型心肌病基因突变与精准医学[J].中华心力衰竭和心肌病杂志,2019(4):231-232.
[2] 陈香璇,程维礼,张郁青,等.TTN及MYH7多基因突变与一家族性扩张型心肌病的相关性分析[J].临床心血管病杂志,2021,37(8):744-748.
[3] Bailly C,Henriques S,Tsabedze N,et al.Role of family history and clinical screening in the identification of families with idiopathic dilated cardiomyopathy in Johannesburg,South Africa[J].S Afr Med J,2019,109(9):673-678.
[4] Cho K W,Lee J,Kim Y.Genetic Variations Leading to Familial Dilated Cardiomyopathy[J].Mol Cells,2016,39(10):722-727.
[5] 周年偉,秦胜梅,刘阳,等.全外显子组测序发现一家族性扩张型心肌病致病基因[J].复旦学报(医学版),2018, 45(2):164-168.
[6] Peters S,Johnson R,Birch S,et al.Familial Dilated Cardiomyopathy[J].Heart Lung Circ,2020,29(4):566-574.
[7] Verdonschot J,Merken JJ,Brunner-La RH,et al.Value of Speckle Tracking-Based Deformation Analysis in Screening Relatives of?Patients With Asymptomatic Dilated Cardiomyopathy[J].JACC Cardiovasc Imaging,2020,13(2 Pt 2):549-558.
[8] Prasad S K,Tayal U.The Value of Strain in Familial Dilated Cardiomyopathy Screening[J].JACC Cardiovasc Imaging,2020,13(2 Pt 2):559-561.
[9] Hirayama-Yamada K,Inagaki N,Hayashi T,et al.A Novel Titin Truncation Variant Linked to Familial Dilated Cardiomyopathy Found in a Japanese Family and Its Functional Analysis in Genome-Edited Model Cells[J].Int Heart J,2021,62(2):359-366.
[10] Li K,Zhao L,Zhang P.Familial dilated cardiomyopathy with a novel LMNA mutation (p.R429C): a case report[J].Cardiol Young,2020,30(10):1544-1546.
[11] de Gonzalo-Calvo D,Quezada M,Campuzano O,et al.Familial dilated cardiomyopathy: A multidisciplinary entity,from basic screening to novel circulating biomarkers[J].Int J Cardiol,2017,228(11):870-880.
[12] Robyns T,Willems R,Van Cleemput J,et al.Whole exome sequencing in a large pedigree with DCM identifies a novel mutation in RBM20[J].Acta Cardiol,2020,75(8):748-753.
[13] Bakalakos A,Ritsatos K,Anastasakis A.Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: a Cardiomyopathy Clinic Doctor's point of view[J].Hellenic J Cardiol,2018,59(5):254-261.
[14] Khayata M,Al-Kindi SG,Oliveira GH.Contemporary char-acteristics and outcomes of adults with familial dilated cardiomyopathy listed for heart transplantation[J].World J Cardiol,2019,11(1):38-46.
[15] McDonagh TA,Metra M,Adamo M,et al.Corrigendum to: 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC[J].Eur Heart J,2021,42(36):3599-3726.
(收稿日期:2022-01-06)
猜你喜欢
现代仪器与医疗(2016年6期)2017-01-12
中国实用医药(2016年29期)2016-12-26
中国实用医药(2016年27期)2016-11-30
农业与技术(2016年15期)2016-11-09
中国实用医药(2016年24期)2016-10-17
中国实用医药(2016年24期)2016-10-17
中国实用医药(2016年13期)2016-07-05
中国实用医药(2016年9期)2016-05-17
现代养生·下半月(2015年9期)2015-09-28
