
匹伐他汀钙及其中间体非对映异构体的合成
2022-05-07 06:12王正泽
药学与临床研究 2022年2期
林 辉,王正泽,王 晓,陈 慧
南京长澳医药科技有限公司,南京 210038
匹伐他汀钙(Pitavastatin Calcium),化学名为(+)-双-[(3R,5S,6E)-7-[2-环丙基-4-(4-氟苯基)-3-喹啉基]-3,5-二羟基-6-烯庚酸]单钙盐,是由日产化学工业株式会社和日本兴和株式会社共同研制的3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,于2003 年首次在日本上市,主要用于高胆固醇血症和家族性高胆固醇血症的治疗。匹伐他汀钙具有肝细胞选择性、半衰期长、药物相互作用少、不影响血糖代谢等优点,市场前景广阔。
目前匹伐他汀钙的商业化生产路线,大部分以母核(3R,5S,6E)-7-[2-环丙基-4-(4-氟苯基)-3-喹啉基]-3,5-二羟基-3,5-O-亚异丙基庚-6-烯酸叔丁酯(2)作为关键中间体或注册起始原料,经两步反应而合成匹伐他汀钙。合成路线见图1。
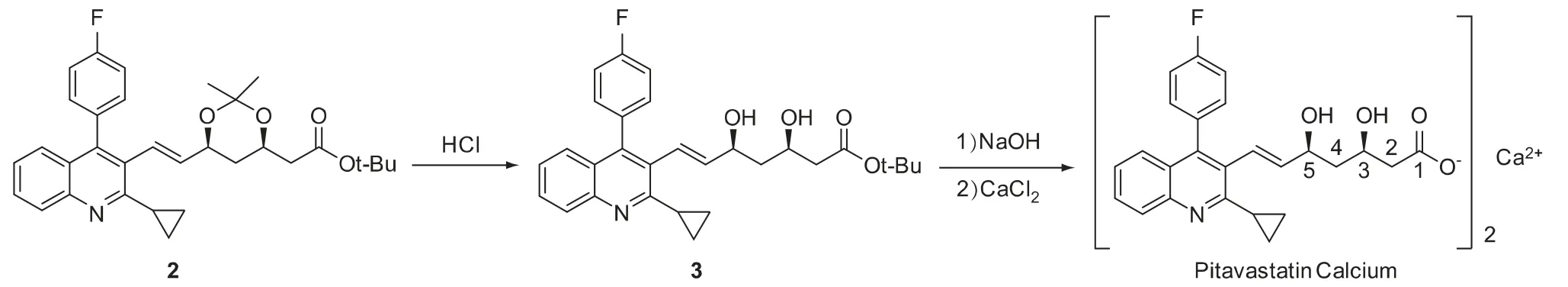
图1 匹伐他汀钙的合成路线
匹伐他汀钙分子中含有两个手性中心,理论上匹伐他汀钙存在一个对映异构体和两个非对映异构体,即C-3 位和C-5 位差向异构体[1]。由于手性药物的不同立体异构体在药效、药代及毒理等方面都可能存在差异,因此手性杂质的控制是非常重要的。由合成工艺可知,匹伐他汀钙手性杂质在由母核2 引入,在后续工艺中,手性碳不是反应位点,在反应中发生翻转的可能性较小。母核2 的手性杂质一旦引入,后续工艺中很难清除,最终转化成产品匹伐他汀钙的异构体杂质。基于源头控制和过程控制的理念,原料药生产企业如选择母核2 作为起始原料或关键中间体,应针对性地建立起手性杂质的内控标准。
手性色谱法可以准确直观地反映各立体异构体杂质的变化情况,是目前用于控制药物手性杂质的主要方法,其前提条件是获得被测杂质的对照品,才能进行专属性和定量限的验证。
现主要研究了匹伐他汀钙非对映异构体中的C-5 位差向异构体(+)-双-[(3R,5R,6E)-7-[2-环丙基-4-(4-氟苯基)-3-喹啉基]-3,5-二羟基-6-烯庚酸]单钙盐(1),以及其关键中间体的C-5 位差向异构体(3R,5R,6E)-7-[2-环丙基-4-(4-氟苯基)-3-喹啉基]-3,5-二羟基-3,5-O-亚异丙基-6-庚烯酸叔丁酯(4)和(3R,5R,6E)-7-[2-环丙基-4-(4-氟苯基)-3-喹啉基]-3,5-二羟基-6-庚烯酸叔丁酯(5)的制备方法。制备上述3 种化合物可以为匹伐他汀钙生产中起始原料的质量控制、中间过程控制和终产品质量标准提供对照品,按照质量源于设计(QbD)的理念,实现对生产工艺的全过程控制。
李纬等[1]首次报道了匹伐他汀钙非对映异构体1 的合成方法,以3-(2-环丙基-4-(4-氟苯基)-3-喹啉基)丙-2-烯醛为原料,经手性辅助剂(R)-(+)-HYTRA 进行不对称羟醛缩合、四甲基三乙酰氧硼氢化铵进行反立体选择性还原、水解成盐而合成化合物1。其中经手性辅助剂(R)-(+)-HYTRA 比较昂贵,其手性纯度比较关键,否则经后续还原终产品中会产生3 个异构体。不对称羟醛缩合反应需要无水无氧和-78 ℃超低温环境,产物也需要柱层析纯化,对照品制备周期较长,无法大规模制备对照品。
本研究在文献报道[2]的基础上,对化合物1 的合成工艺进行了改进,以化合物2 为原料,经盐酸脱缩酮、二氧化锰氧化、四甲基三乙酰氧硼氢化铵进行反立体选择性还原,得到化合物5,再经水解成盐得到化合物1。参考文献[3]将化合物5 与2,2-二甲氧基丙烷在对甲苯磺酸的催化下,形成O,O-缩酮得到化合物4。合成路线见图2。
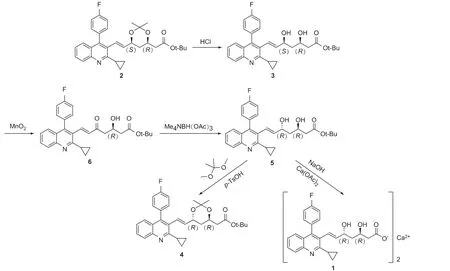
图2 匹伐他汀钙及中间体C-5 位差向异构体的合成
化合物1 的合成,选择市场容易获得且手性纯度较高的化合物2 作为原料,保证了C-3 位碳原子的构型为R。在氧化反应中,选择了活性二氧化锰作为催化剂,反应转化率高,后处理简单,无需柱层析纯化产品。化合物6 使用四甲基三乙酰氧硼氢化铵进行反立体选择性还原,产物经乙酸乙酯/正己烷精制后得(3R,5R)∶(3R,5S)两种构型,比例为98.6∶1.4。
1 仪器与药品、试剂
1.1 仪器
U3000 型高效液相色谱仪(美国UltiMate 公司);7890 型高效液相色谱仪(美国Agilent 公司);AV-500 型核磁共振仪(德国Bruker 公司);6200 Q-TOF 型质谱仪(美国Agilent 公司);SGW-2 型自动旋光仪(上海精密科学仪器有限公司)。
1.2 药品与试剂
柱层析使用100~200 目硅胶(青岛海洋化工五厂,试剂级);化合物2(北京海美桐医药科技有限公司,含量99%);活性二氧化锰(成都艾科达化学试剂有限公司,含量80%);四甲基三乙酰氧硼氢化铵(成都吉安特医药科技有限公司,含量98%);其他试剂均为市售分析纯;水为重蒸馏水。
2 试验方法
2.1 化合物3 的合成
将2(30.0 g,58.0 mmol)加入乙腈(300 mL)中,搅拌均匀。滴加1 mol·L-1盐酸(63.8 mL,63.8 mmol),升温至40 ℃,搅拌2 h。加入水(200 mL)稀释,饱和碳酸氢钠溶液调节pH 7,乙酸乙酯(200 mL)萃取。有机层用饱和NaCl 溶液(100 mL)洗涤,经无水硫酸钠干燥后过滤,滤液减压蒸除溶剂。固体物用乙酸乙酯/正己烷重结晶,得白色固体化合物3(26.1 g,以2 计收率94.2%)。[α]20D=+45.0°(c=1.0,MeOH);1H-NMR(500 MHz,CDCl3)δ:7.95 (d,J=8.5 Hz,1H,Ar-H),7.55~7.59(m,1H,Ar-H),7.30~7.34(m,2H,Ar-H),7.15~7.29(m,4H,Ar-H),6.64(d,J=16.5 Hz,1H,=CH),5.61(dd,J=6.0,16.0 Hz,1H,=CH),4.40(m,1H,-CH),4.08(m,1H,-CH),3.71(br,1H,-OH),3.30(br,1H,-OH),2.44(m,1H,-CH2),2.34~2.40(m,2H,-CH2,-CH),1.44~1.49(m,10H,-CH2,-CH3),1.28~1.34(m,3H,-CH2),1.01~1.03(m,2H,-CH2);HRMS-ESI calcd for C29H33FNO4[M+H]+478.2388,found 478.2393。
2.2 化合物6 的合成
将3(25.0 g,52.34 mmol)溶于干燥二氯甲烷(250mL)中,分批加入活性二氧化锰(68.5g,0.63mol),回流搅拌6~8 h。抽滤,滤饼用二氯甲烷(50 mL×3)洗涤。合并滤液,滤液减压蒸除溶剂,得黄色油状化合物6(23.8 g,以3 计收率95.7%)。[α]20D=+14.1°(c=1.0,MeOH);1H-NMR(500 MHz,CDCl3)δ:7.97(d,J=8.5 Hz,1H,Ar-H),7.62~7.68(m,2H,=CH),7.32~7.37(m,2H,Ar-H),7.19~7.26(m,4H,Ar-H),6.36(d,J=16.5 Hz,1H,=CH),4.39(br,1H,-OH),3.45(s,1H,-CH),2.62~2.72(m,2H,-CH2),2.40~2.42(m,2H,-CH2),2.32~2.36(m,1H,-CH),1.46(s,9H,-CH3),1.39~1.41(m,2H,-CH2),1.093(m,2H,-CH2);MSESI m/z:476.1[M+H]+。
2.3 化合物5 的合成
依次将乙酸(240 mL)和四甲基三乙酰氧硼氢化铵(92.1 g,0.35 mol)溶于乙腈(240 mL)中,室温搅拌2 h。冷却至-20 ℃,将6(23.8 g,50.08 mmol)溶于乙腈(240 mL)和乙酸(80 mL)中,缓慢滴加到反应液中,继续搅拌4 h。将反应液加入饱和碳酸氢钠溶液(500 mL)淬灭,乙酸乙酯(300 mL×2)萃取。合并有机层,饱和NaCl 溶液(150 mL×2)洗涤,经无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,得浅棕色油状物。该物用乙酸乙酯-正己烷重结晶,得浅黄色固体5(18.6 g,以6 计收率77.8%)。纯度98.6%[HPLC 归一化法:色谱柱OD-H(4.6 mm×250 mm,5 μm);流动相为正己烷-乙醇(95∶5);检测波长245 nm;柱温30 ℃;流速1.0 mL·min-1。相对保留时间0.9(相对于化合物3)]。[α]20D=+8.9°(c=1.0,MeOH);1H-NMR(500 MHz,CDCl3)δ:8.09(br,1H,Ar-H),7.62~7.65(m,1H,Ar-H),7.34~7.39(m,2H,Ar-H),7.20~7.31(m,4H,Ar-H),6.74(d,J=16.15 Hz,1H,=CH),5.73(dd,J=5.3,16.15 Hz,1H,=CH),4.48(s,1H,-CH),4.07(s,1H,-CH),3.63(br,1H,-OH),3.15(br,1H,-OH),2.44~2.51(m,1H,-CH),2.32~2.41(m,2H,-CH2),1.65~1.70(m,1H,-CH2),1.51~1.52(m,9H,-CH3),1.41~1.46(m,3H,-CH2),1.10~1.11(m,2H,-CH2);HRMS-ESI calcd for C29H33FNO4[M+H]+478.2388,found 478.2401。
2.4 化合物1 的合成
将5(10.0 g,20.94 mmol)溶于无水乙醇(60 mL)中,加入1 mol·L-1氢氧化钠(25.1 mL,25.1 mmol),室温搅拌1 h,减压蒸除乙醇。固体物加入水(140 mL)稀释,用0.1 mol·L-1盐酸调节pH 8。冰浴冷却,滴加1 mol·L-1乙酸钙(14.7 mL,14.66 mmol),搅拌3 h,过滤,滤饼水洗涤,烘干,得浅黄色固体1(8.9 g,以5计收率96.7%)。纯度98.11%[HPLC 归一化法:色谱柱C18(4.6 mm×250 mm,5 μm);流动相A 为pH3.8醋酸钠缓冲溶液,流动相B 乙腈,梯度洗脱条件为:0→20 min:A 为60%,20→40 min:A 为60%→30%,40→65 min:A 为30%。检测波长245 nm;柱温40℃;流速1.0 mL·min-1。相对保留时间1.1(相对于匹伐他汀钙)]。[α]20D=-0.4°(c=1.0,CH3CN/H2O=1/1)[文献[1]:[α]20D=-0.4°(c=1.0)];1H-NMR(500 MHz,DMSO-d6)δ:7.87(d,J=8.35 Hz,1H,Ar-H),7.64(t,J=7.65 Hz,1H,Ar-H),7.27~7.39(m,6H,Ar-H),6.52(d,J=16.1 Hz,1H,=CH),5.665(br,1H,-OH),5.62(dd,J=5.15,16.15 Hz,1H,=CH),4.88(br,1H,-OH),4.20(s,1H,-CH),3.91(s,1H,-CH),2.55(m,1H,-CH),2.12(dd,J=8.9,15.35 Hz,1H,-CH2),2.02(q,J=8.9 Hz,1H,-CH2),1.15~1.22(m,4H,-CH2),1.03~1.05(m,2H,-CH2);13C-NMR(125 MHz,DMSO-d6)δ:178.82,162.59,160.59,145.59,143.65,142.12,133.10,132.07,131.92,129.73,128.74,128.36,125.65,125.55,122.51,115.29,115.12,67.46,44.42,44.14,15.36,10.78;HRMS-ESI calcd for C25H25FNO4[M+H]+422.1762,found 422.1769.
2.5 化合物4 的合成
将5(8.5 g,17.8 mmol)溶于2,2-二甲氧基丙烷(85 mL)中,加入一水对甲苯磺酸(0.85 g),室温搅6 h,减压蒸干。固体物经硅胶柱色谱[洗脱液:正己烷:乙酸乙酯(10∶1)]纯化,得浅黄色固体4(7.3 g,以5 计收率79.3%)。纯度97.6%[HPLC 归一化法:色谱柱C18(4.6 mm×250 mm,5 μm);磷酸二氢钾溶液(pH 6.8)-乙腈(35∶65)。检测波长245 nm;柱温40 ℃;流速1.0 mL·min-1。相对保留时间1.1(相对于化合物2)]。1H-NMR(500 MHz,CDCl3)δ:7.96(s,1H,Ar-H),7.55~7.60(m,1H,Ar-H),7.27~7.41(m,2H,Ar-H),7.05~7.25(m,4H,Ar-H),6.53(d,J=16.3 Hz,1H,=CH),5.69(dd,J=6.05,16.25 Hz,1H,=CH),4.33(q,J=7.05 Hz,1H,-CH),4.14~4.19(m,1H,-CH),2.38~2.44(m,2H,-CH2,-CH),2.33(dd,J=5.5,15.1 Hz,1H,-CH2),1.48~1.58(m,2H,-CH2),1.44(s,9H,-CH3),1.33~1.34(m,8H,-CH2,-CH3),1.04~1.05(m,2H,-CH2);13C-NMR(125 MHz,DMSO-d6)δ:169.98,163.25,161.73,160.67,141.35,137.90,135.22,133.14,131.97,128.81,128.46,125.99,125.43,115.27,100.40,80.58,67.73,63.47,42.20,36.77,28.08,25.55,24.73,15.92,10.50;MS-ESI m/z:540.3[M+Na]+。
3 结论
本研究成功地合成了匹伐他汀钙及其中间体非对映异构体中的C-5 位差向异构体,并经核磁共振氢谱、碳谱、质谱和比旋光度等确证结构,3 个目标化合物5、1 和4 的总收率分别为70%、68%和56%(以2 计),纯度经HPLC 法检测均在97.5%以上,可以作为匹伐他汀钙原料药及其中间体质量控制的对照品。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
读与写·下旬刊(2018年6期)2018-07-14
山东工业技术(2018年9期)2018-05-26
科学与财富(2017年17期)2017-06-16
电子技术与软件工程(2016年24期)2017-02-23
化学教学(2015年12期)2015-12-12
股市动态分析(2015年12期)2015-09-10
中国医药科学(2015年4期)2015-05-20
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
新媒体研究(2014年10期)2014-06-26
