
老年高度复杂异常染色体核型MDS误诊为ITP 1例
2022-04-28 11:14邢明泉葛洪峰吴维霞孙晓星马兰
老年医学研究 2022年2期
邢明泉,葛洪峰,吴维霞,孙晓星,马兰
1亳州市人民医院血液内科,安徽亳州 236800;2亳州市人民医院影像中心,安徽亳州 236800;3亳州市人民医院感染内科,安徽亳州 236800
原发免疫性血小板减少症(ITP)属自身免疫性疾病范畴,可见于任何年龄段,主要临床特征为由免疫因素诱发的以单系血小板减少而引起的以出血为主要表现的血液系统疾病,临床可出现皮肤黏膜出血、牙龈渗血、鼻腔出血、呕血、咯血,甚至脑出血,骨髓形态学巨核细胞增多为常见特征[1]。ITP的成人年发病率大约为6/10万,65岁以上老年人群及4岁以下儿童发病率相对较高,女性较男性发病率高[2]。ITP治疗的主要目标是尽量减少出血事件,在此基础上提高血小板水平[3]。骨髓增生异常综合征(MDS)是一组来源于骨髓造血干细胞,以髓系异常克隆性增殖分化伴发育异常为主要特征的一类异质性较大的疾病。骨髓一系或多系病态造血致外周血细胞一系或多系减少,存在向急性髓系白血病(AML)转化的风险[4]。临床主要特征为外周血一系或者多系血细胞减少引起的感染、出血、难治性贫血等。主要发病群体为老年人,中位发病年龄为70岁[5]。MDS相关基因突变及异常的染色体核型是致MDS发生的始动因素,因此MDS往往存在多种基因突变及异常染色体核型,高度复杂异常染色体核型是其中的一种类型,属于MDS高危预后因素,临床发生率低,但预后极差[6]。高度复杂异常染色体核型MDS临床症状及骨髓形态学可不典型,易被漏诊或误诊,本文报告了1例老年患者误诊为ITP的整个过程,总结误诊经验,为临床诊疗此类疾病提供依据。
1 病例资料
1.1 基本资料 患者男,72岁,2021年1月19日因“咯血4 d,发现血小板减少1 d”入住亳州市人民医院血液内科。患者4 d前无明显诱因出现咯血,无鼻腔出血及牙龈出血,无皮肤瘀点瘀斑,无发热,无呕血,无黑便,无血尿,余无特殊不适主诉,遂就诊于当地卫生院。检查血常规:白细胞4.27×109/L,血红蛋白98.0 g/L,血小板6×109/L。胸部CT:两肺多发高密度影,考虑感染可能。患者因血小板重度减少入我科进一步治疗。患者既往体健,无糖尿病、高血压等慢性病,无乙肝、结核等传染性疾病,无肿瘤家族史。
1.2 入院查体及实验室检查 患者体温正常,神志清,精神差,贫血貌,皮肤黏膜无瘀点瘀斑,全身浅表淋巴结未触及肿大,肝、脾未触及,胸骨压痛阴性,心肺听诊未见明显异常。血常规:白细胞3.74×109/L,血红蛋白95.0 g/L,血小板12×109/L,超敏C反应蛋白52.20 mg/L,自身抗体16项均为阴性、肝肾功能及贫血四项均无异常,腹部彩超未发现肝脾肿大。
1.3 诊疗经过 结合患者临床表现及实验室检查考虑诊断为ITP可能。入院次日患者出现发热,最高体温38.9℃,无寒战畏寒,仍有咯血症状。请感染科会诊考虑肺部感染,结核不除外。遵会诊意见完善抗酸杆菌检查,结核杆菌DNA及多次结核菌痰培养均为阴性,痰细菌学培养及痰涂片查真菌均无病原体阳性结果。入院次日即完善骨髓穿刺及活检检查。第3天,骨髓形态学检查结果回报显示:骨髓增生明显活跃,未见明显发育异常,骨髓巨核细胞增多。骨髓活检病理(合肥千麦医学检验实验室,病理号G2100875):造血组织中,粒系及中晚幼及以上阶段为主,幼红细胞比例增高,无明显病态造血(图1)。免疫分型(合肥千麦医学检验实验室,编号210103979):淋巴细胞约占有核细胞的16%,分布大致正常;单核细胞约占有核细胞的7.5%,比例偏高,表型成熟,粒细胞约占有核细胞的50%,未见明显发育异常(图2)。查胸部CT:两肺多发渗出性改变,考虑感染或出血可能。综合以上考虑ITP、肺出血渗出伴感染可能、咯血。治疗上予地塞米松联合丙种球蛋白冲击治疗,头孢哌酮舒巴坦联合甲磺酸左氧氟沙星抗感染治疗。患者咯血症状无明显好转,予血小板1个治疗量输注,复查血小板24×109/L,继续予激素联合丙种球蛋白冲击治疗,抗感染及止血治疗。患者发热、咯血等症状好转。大约2周后,患者骨髓染色体核型分析(合肥千麦医学检验实验室,编号 K53039)检验结果:46,XY,del(5)(q15q33)[5]/46,idem,del(6)(q13),del(6)(q25),add(16)(q13),add(17)(q23),add(18)(q21)[3]/68~83〈4n〉,XXY,-Y,-2,-3,del(5)(q15q33)×2,-9,-?10,-12,-13,-15,-16,add(16)(q13),-17,-18[cp7]/46,XY[5],提示:此患者标本经培养后分析20个中期相细胞,15个细胞核型存在染色体畸变,其中7个细胞核型表现为四倍体复合核型(图3)。结合染色体核型结果,根据2019年《骨髓增生异常综合征诊断与治疗专家共识》[7],该患者修订诊断为MDS,高度复杂异常染色体核型,IPSS评分:中危-2,IPSS-R评分:高危。
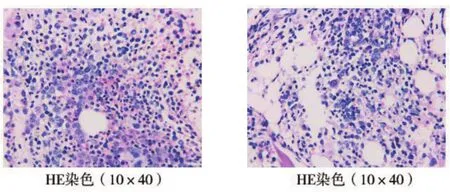
图1 骨髓病理检查
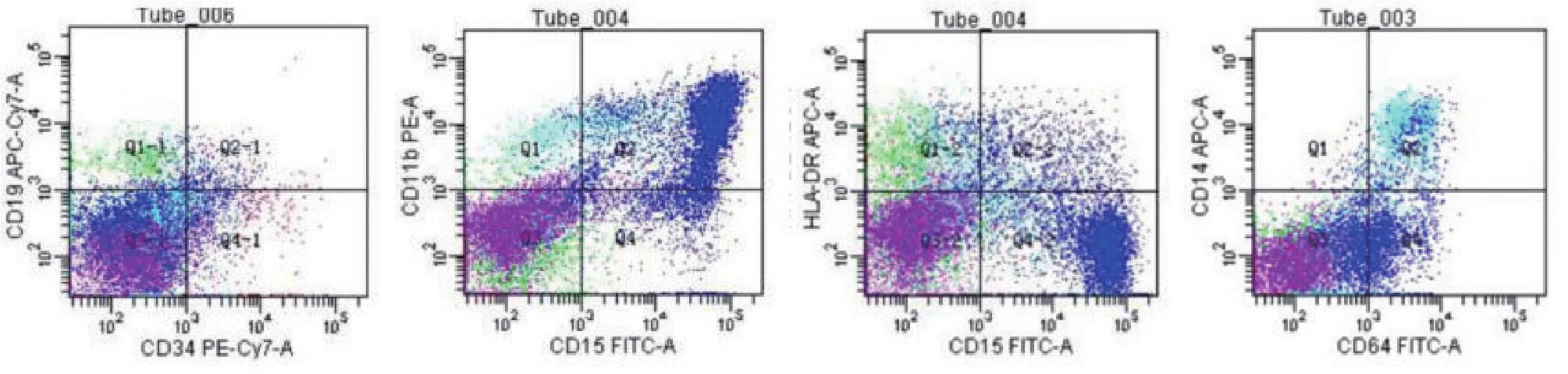
图2 免疫分型
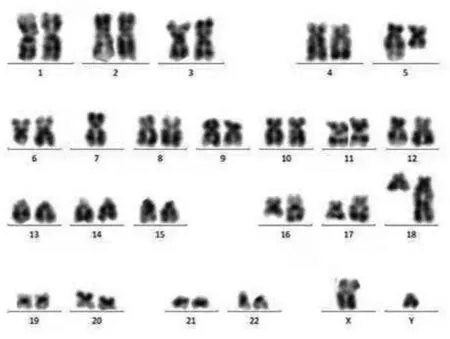
图3 骨髓染色体核型分析
患者修订诊断为MDS后遂予停激素及丙种球蛋白,予沙利度胺调节免疫,雄激素促造血等治疗,患者咯血症状逐渐好转,未再发热。复查血常规,血小板升高至30×109/L,考虑治疗有效。患者为高度复杂异常染色体核型,建议患者尽快启动去甲基化治疗,因恰逢春节,患者及家属要求春节后行去甲基化治疗,遂出院。患者于2021年2月10日下午无明显诱因突发胸闷,伴心慌气促明显,遂打120急诊入住我院急诊内科,测血氧饱和度为45%,心率、血压可,心电监护下推入抢救室,患者经人工气囊应用,呼吸机支持治疗均无效,于当天夜里死亡。患者死亡直接原因为呼吸衰竭,根本原因为基础疾病进展。
2 讨论
患者主因咯血入院,入院血常规检查示白细胞、血红蛋白均无明显减少,血小板为12×109/L,余检查无明显异常,排除结核感染后,考虑肺出血渗出伴感染可能,予抗生素治疗。后骨髓形态报告显示骨髓增生活跃,未见明显异常造血,巨核细胞增多。免疫分型提示粒系及单核系无明显分化异常,骨髓病理也无明显病态造血。患者肝脾无肿大,结合患者临床特征及检查很难联想到MDS诊断,所以当时主要考虑ITP合并肺出血伴感染可能,治疗上予激素联合丙种球蛋白冲击治疗。因骨髓染色体培养需要较长时间,有时候会有培养不出染色体等可能,所以染色体报告发布往往较迟。骨髓形态、免疫分型、骨髓病理等均无MDS相关特征,且外周血仅以血小板减少为主要表现时,很不容易考虑MDS诊断,而该患者骨髓形态又出现巨核细胞增多,更加提示ITP可能。患者有咯血症状,按照国内外专家共识,为迅速止血预防患者重要脏器出血,遂启动ITP相关治疗。约2周后患者骨髓染色体核型分析结果显示为非常复杂染色体核型,遂即修订诊断为MDS高危 高度复杂异常染色体核型。予停激素及丙种球蛋白,予沙利度胺调节免疫,雄激素促造血治疗,建议患者尽快去甲基化治疗,但因春节原因患者要求节后启动去甲基化治疗。出院后患者无明显诱因下突发胸闷,抢救无效死亡。再次回顾其死亡原因,基础疾病是根本原因。系统回顾该患者的诊治过程,患者临床症状及相关检查具有极强的干扰性,在其骨髓染色体核型结果未出报告之前,虽然ITP诊断缺乏特异性,属于排除性诊断,但当外周血出现以单纯性血小板减少为主,无明显肝脾肿大,骨髓形态学及免疫分型无病态造血和分化异常的情况下巨核细胞增多,临床最容易考虑到的首先是ITP诊断,很难考虑到MDS,临床极易发生误诊误治。
ITP是异常免疫介导的复杂的异质性疾病,骨髓形态学主要特征为骨髓巨核细胞成熟障碍及巨核细胞增多,临床诊断ITP往往无金标准,依据排除性诊断,该患者外周血单纯以血小板减少并以咯血为主要表现,无明显肝脾肿大,骨髓形态学及免疫分型无病态造血和分化异常,骨髓巨核细胞增多,故考虑ITP诊断。根据国内外专家最新共识,其治疗目标主要是尽量减少出血和相关并发症,因此为预防患者发生严重的出血性疾病遂启动激素联合丙种球蛋白治疗。有研究表明仅20%左右的的成人ITP患者可以达到自发性缓解,约80%患者进展成慢性ITP[8]。成人ITP治疗首选糖皮质激素,其反应时间短、有效率高。2019版ASH指南建议,对于外周血血小板计数小于或等于30×109/L,无论有无出血症状均优先推荐使用大剂量地塞米松冲击治疗[9]。丙种球蛋白可以迅速升高血小板,临床上用于预防严重出血以及对糖皮质激素不耐受的ITP患者[10],临床安全有效,有效率可高达80%[11],且具有良好的临床耐受性。因此对于初诊ITP、血小板严重减少伴出血明显的患者临床常常应用激素联合丙种球蛋白来迅速升高血小板,预防出血症状加重。对初始激素治疗无效或复发的成人ITP,建议行以脾切除、利妥昔单抗、血小板生成素受体激动剂等二线治疗方案[12-13]。ITP不同的病程对其预后有较大影响,越早治疗,药物反应越好,预后越好,反之越差[14]。因此结合患者临床特征及辅助检查,按照国内外最新专家共识,治疗的选择是正确的。
而MDS是一组临床特征、形态学特征、细胞遗传学特征、基因特征等均有明显异质性的骨髓异常克隆性疾病[15]。MDS异质性较大,骨髓病态造血致外周血一系及多系血细胞减少为MDS主要临床特征[16],基因突变及染色体核型异常为其遗传学特征。MDS患者染色体核型异常可以作为MDS危险分层的重要依据,其与MDS治疗及预后密切相关[17]。有研究报道其染色体核型异常的发生率约为40%[18]。常见的异常染色体类型为染色体缺失、单体或三体,按照发生异常的概率依次为5号染色体异常、8号染色体异常、7号染色体异常、20q-,Gangat等异常核型。在评价MDS预后的IPSS和IPSS-R积分系统中,染色体异常检出率越高,危险度越高[19]。有研究表明,异常染色体核型MDS更易发生难治的血小板减少或难治性贫血,复杂异常染色体核型更易发生全血细胞减少,因此染色体核型分析对这部分患者更具诊断价值,更能预测患者预后[20]。国外有研究表明,染色体核型异常可以先于骨髓病态造血,即在MDS患者血细胞无明显下降甚至下降不明显时,染色体就已经发生了畸变,尤其是复杂异常核型往往是直接导致患者较差预后的关键因素[21]。该患者可能是染色体异常核型发生在病态造血之前,在患者无明显临床症状时不易被发现,当患者出现严重血小板减少时才使得患者就医。复杂异常染色体核型MDS具有较短的生存期,中位生存时间较短,早期病死率较高,有着更高的向急性白血病转化风险。因此复杂异常染色体核型为MDS患者预后不良的重要因素之一。研究表明3个染色体异常的复杂核型有着较短的生存期,大于3个染色体异常的复杂核型生存期会更短[22]。本例患者为高度复杂异常染色体核型,共分析20个中期相细胞,15个细胞核型存在染色体畸变,其中7个细胞核型表现为四倍体复合核型,临床少见。患者从出现临床症状到死亡不足两个月,生存期极短,与文献报道相一致,如果这类患者早些启动MDS相关治疗会不会改善患者的预后,值得临床进一步研究。
综上,本例患者临床表现及骨髓形态病理学等均较符合ITP,后骨髓染色体核型分析为高度复杂异常染色体核型,遂修订诊断为MDS。这类MDS临床症状及骨髓形态病理学等非常不典型,因骨髓染色体培养时间较长或者常规培养不敏感,因此早期极易发生误诊误治,而复杂染色体核型预后极差,越早治疗预后相对越好,临床需及时追踪染色体结果,必要时选择更加敏感的染色体检查方法,如染色体芯片检查,对于可疑患者加做MDS高频突变基因检查,以免误诊误治延误病情。
猜你喜欢
妇女生活(2022年5期)2022-06-09
中国典型病例大全(2022年7期)2022-04-22
健康体检与管理(2022年2期)2022-04-15
青年文学家(2020年16期)2020-07-13
文苑(2018年18期)2018-11-08
健康博览(2018年1期)2018-10-23
中国实用医药(2016年36期)2017-06-20
医学信息(2016年29期)2016-11-28
诗林(2016年5期)2016-10-25
湖北农业科学(2015年17期)2015-10-09
