
Role of hydrogen in the dissociation of CH4 on different graphene by DFT study
2022-04-27 08:21KunLIHejunLINiLIQingSONGLehuQI
Chinese Journal of Aeronautics 2022年6期
Kun LI, Hejun LI,*, Ni LI, Qing SONG, Lehu QI
a State Key Laboratory of Solidification Processing, Carbon/Carbon Composites Research Center, Northwestern Polytechnical University, Xi’an 710072, China
b School of Aeronautics, Northwestern Polytechnical University, Xi’an 710072, China
c School of Mechanical Engineering, Northwestern Polytechnical University, Xi’an 710072, China
KEYWORDS Abstraction;Dehydrogenation;DFT;Graphene;Hydrogen;Methane
Abstract The effect of hydrogen on the growth mechanism of pyrocarbon has attracted much attention. The influence of hydrogen on the dissociation from CH4 to C2H2 on pristine graphene,N-doped graphene and vacancy graphene have been investigated by using density functional theory.There are two kinds of heterogeneous reaction pathways when the hydrogen is involved,i.e.,dehydrogenation reactions and H-abstraction reactions. The transition state calculations were performed to acquire the reaction pathways on each substrate after obtaining the most stable adsorption configurations of the reactants and products. The results indicate that the adsorptions of reactants are not affected by hydrogen. The dehydrogenation reactions are more favored on vacancy graphene, and the H-abstraction reactions are more favored on the pristine graphene and N-doped graphene.The dehydrogenation reactions on the vacancy graphene are most favored among all these reactions in favor of the deposition of pyrocarbon.The dehydrogenation reactions on these three substrates are affected by hydrogen.
1. Introduction
Carbon fiber reinforced carbon matrix (C/C) composites have been widely used as high-temperature hot end-component in the aeronautic industry owing to their excellent thermal and mechanical properties, like high strength and stiffness, high thermal conductivity, low thermal expansion and ablation resistance.Chemical Vapor Infiltration (CVI) using methane as carbon precursor represents a popular approach to fabricate the high-performance and low-cost C/C composites.During the CVI process, the formation of soot and coke would significantly reduce the density of C/C composites and different textures of pyrocarbon would lead to different application performances,and this suggests the importance and necessity of producing high-quality C/C composites,which is crucial for both fabrication application and scientific research.
Up to now, many researchers have been studying the factors for controlling the formation and growth mechanisms of pyrocarbon.Wang et al.compared the effects of Thermal Chemical Vapor Infiltration (TCVI) and Isothermal Chemical Vapor Infiltration (ICVI) on the textures of pyrocarbon, and concluded that the high-textured pyrocarbon of ICVI was uniform and showed a more distinct long-range order than that of TCVI. Dong and Hu¨ttingerdeclared that the high-textured pyrocarbon could be produced by controlling the ratio of aromatic hydrocarbons to small linear hydrocarbons during the CVI process. In the meanwhile, the effect of hydrogen on the growth mechanism of pyrocarbon has attracted much attention.Benzinger and Hu¨ttingeranalyzed the CVI process of pyrocarbon with respect to hydrogen that is regarded as both a reaction product and a non-inert diluent gas, and identified strategies that could contribute to the optimization of the CVI process. Becker and Hu¨ttingerstudied the influence of hydrogen on the pyrocarbon deposition from CHin the low-temperature regime, and revealed that hydrogen showed a clear inhibiting effect on the formation of CHand CHand the pyrocarbon deposition rate decreased with the increase of hydrogen partial pressure. Ziegler-Devin et al.also proved the obvious inhibiting effect of hydrogen on the formation of pyrocarbon and aromatic species. To the best of our knowledge, little attention has been paid to the influence of the hydrogen on the dissociation of CHduring the CVI process of C/C composites. It is known that the dissociation of carbon source (CH) is the precondition for the formation of pyrocarbon, and the dissociation products would directly affect the growth mechanism of pyrocarbon.Therefore, to explore how hydrogen influence and change the quality of C/C composites, it is essential to figure out the role of hydrogen in the dissociation of CHfor the production of C/C composites.
During the past two decades, graphene has been widely used as the ideal microstructure model for graphite, coal, carbon black,pyrocarbon and many carbon-based materials,and the dissociation of CHon substrate surfaces like graphene and metalhave been reported by researchers.In this study, graphene is used as the surface of pyrocarbon and carbon fibers since the atomic structures of the carbon fibers and the deposited pyrocarbon are both turbostratic graphite structure.To make the simulation substrate model get closer to the real experiment one, the N-doped graphene and vacancy graphene are introduced to this study. Because nitrogen element is commonly observed in the carbon fiber of C/C compositesand vacancy defect is a typical defect widely spread in the C/C composites,the N-doped graphene and vacancy graphene can be used as the substrate here.Besides, the dissociation of CHare reported to happen on the surfaces of carbon fiber and pyrocarbon during the CVI process, so the dissociation mechanisms on the pristine graphene, N-doped graphene and vacancy graphene are studied to explore how different graphene substrates influence the dissociation mechanisms. Also, because the reactions are very numerous and complicated during the dissociation of CHfor the production of C/C composites,we only consider the dissociation from CHto CHfor simplicity and convenience. Thus, in this work, a systematic study is performed to investigate the reaction mechanisms of the dissociation from CHto CHon the H-adsorbed pristine graphene, N-doped graphene and vacancy graphene.
2. Computational method
2.1. Model
According to the previous studies,there are two kinds of heterogeneous reaction pathways when hydrogen is involved, i.e. direct dehydrogenation reactions and Habstraction reactions.In the direct dehydrogenation reactions,a C-H bond is decomposed and another new H radical is formed; in the H-abstraction reactions, an existing H radical abstracts a hydrogen atom from a C-H bond and forms a hydrogen molecule. And the primary heterogeneous reaction pathways from methane to acetylene in pyrocarbon deposition can be concluded as
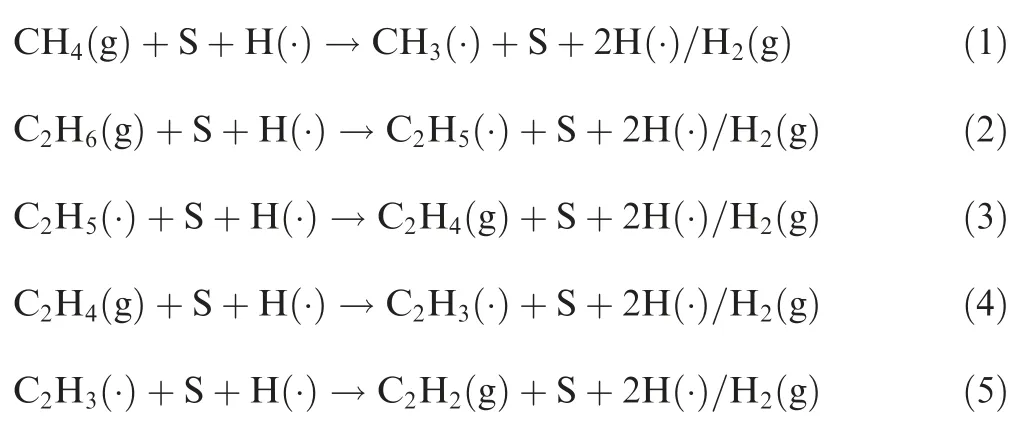
where S represents the substrates (i.e., pristine graphene, Ndoped graphene, or vacancy graphene); (g) represents hydrocarbon gas; (·) represents radical. As can be seen from Reactions (1)-(5), the formation reaction of CHfrom CH(2CH→CH) is not studied since there is neither direct dehydrogenation step nor H-abstraction step happened to it.The optimized structures of pristine graphene, N-doped graphene and vacancy graphene are shown in Fig.1,respectively,and the bond lengths are plotted.The pristine graphene is represented by a periodic single layer graphene that contains 50 carbon atoms, and the lattice parameters in a, b and c directions are separately set to be 12.30 A° , 12.30 A° and 20.20 A°(1 A° =10m).The N-doped graphene is modelled by replacing one carbon atom of pristine graphene with a nitrogen atom, and the vacancy graphene is obtained by removing one carbon atom of pristine graphene. These three models are calculated to be big enough to avoid the interactions among the defects and the adsorbates in the adjacent cells.And graphene is usually produced by chemical vapor deposition and it is therefore believed to be stable as the substrate.The H coverage is not considered, and we only explore the influence of single H radical on the dissociation reactions mechanisms. For both physisorption and chemisorption, the adsorption energy Eis defined as

where Eis the energy of the optimal graphene; Eis the energy of the optimal adsorbate; Eis the total relaxed energy of the adsorption process at the equilibrium state.
2.2. Method
The electron change and correlation interactions were described by the Generalized Gradient Approximation(GGA) with Perdew-Burke-Ernzerhof (PBE) functional,and the all-electron core treatment and double-numerical basis set with polarization functions (DNP) which is the same as 6-
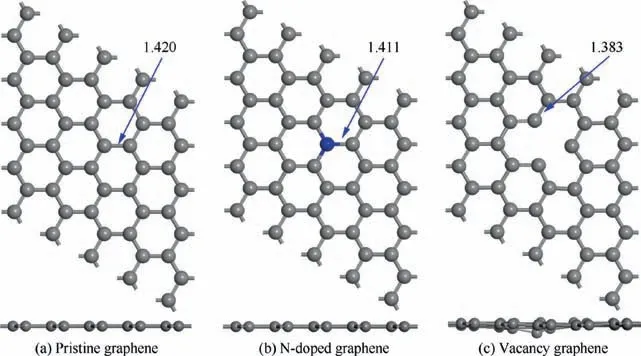
Fig. 1 Top view and side view of the most stable optimized structures of pristine graphene, N-doped graphene and vacancy graphene.
31G** basis set were selected for all atoms.The van der Waals forces were handled by the Density Functional Theory + Dispersion (DFT + D) method within the Grimme scheme.The methods employed in this DFT calculation are widely used in many publications for its reliability and accuracy in different fields,such as the adsorption of molecules like methane on graphene,the dehydrogenation of molecules like propane and ammonia on graphene,and the dehydrogenation of hydrocarbons on metal surfaces.Besides, the bond lengths of these substrates marked in
Fig. 1 are consistent with the experimental results, which can help justify the credibility of the methods. To investigate the Transition State (TS) and the minimum energy pathway for the dissociation from CHto CHon H-adsorbed pristine graphene, N-doped graphene and vacancy graphene, the Linear Synchronous Transit/Quadratic Synchronous Transit(LST/QST) and Nudged Elastic Band (NEB) tools were used.After obtaining the TS structure of each reaction, the energy barrier and reaction energy can be obtained as

where Eis the energy barrier of each reaction; Eis the energy of the TS structure; Eis the energy of each reactant;Eis the reaction energy of each reaction; Eis the energy of each product. The other detailed parameters used in the simulation were summarized in Table 1.The calculations were performed using the Dmolcodeembedded in the Material Studio package (Accelrys, SanDiego, CA).
3. Results and discussion
3.1. Adsorption of H on pristine graphene, N-doped graphene and vacancy graphene
Before investigating the role of hydrogen in the·dissociation of CHon different graphene substrates, the behavior of H adsorbed on pristine graphene, N-doped graphene and vacancy graphene are discussed. After testing all the possible adsorption configurations, the most stable ones are depicted in Fig. A1. As can be seen from Table A1, these adsorptionstructures are equipped with large adsorption energies, which demonstrates that these interactions are all strong chemisorption which is different from the physisorption of hydrogen molecule on the substrates. To further understand the mechanisms of these interactions, the electronic properties of the three H-adsorbed substrates are also analyzed. The charge density differences of the three H-adsorbed substrates are calculated to illustrate the electron transfer during the adsorption,and the top view and side view of the charge density differences are presented in Fig. 2. The area shaded in blue indicates charge depletion, whereas that shaded in the yellow area indicates charge accumulation. As indicated in Fig. 2, there is a significant overlap of electron clouds between the H atoms and the substrates in these configurations,and all the H atoms here donate their electron to the substrates to strengthen their chemisorption. The charge depletion of the H atom can be attributed to its smaller electronegativity than that of the C atom.Besides,the blue region of the H-adsorbed vacancy graphene is the largest among the three substrates,and its adsorption energy is also the largest. This phenomenon proves that the vacancy graphene is more active than the other two substrates,which can be attributed to the unbonded carbon atoms in the vacancy.
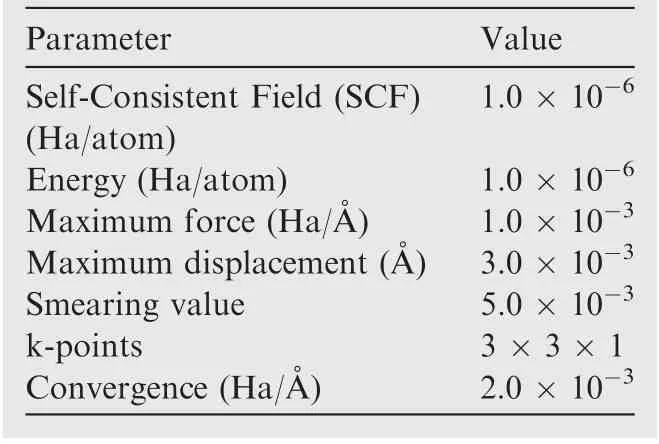
Table 1 Parameters used in DFT calculation.
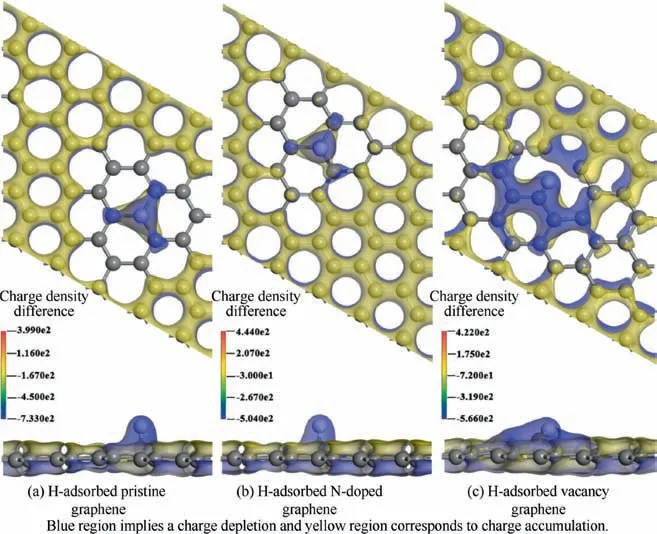
Fig. 2 Top view and side view of charge density differences of pristine graphene, N-doped graphene and vacancy graphene.
Furthermore, the Density of States (DOS) of these substrates is studied and exhibited in Fig. 3, and the densities are compared with the DOS of the substrates without the adsorbed H atom. As can be observed from Fig. 3(a), after the adsorption of the H atom the electronic peak near the Fermi level is noticeably increased while the rest parts remain nearly the same.And the change near the Fermi level is mainly caused by the adsorbed H atom,which can be explained by the Partial Density of States (PDOS) of the H-adsorbed pristine graphene in Fig. A2. In Fig. 3(b), the DOS of H-adsorbed N-doped graphene is moved towards right around 1 eV when compared with that of N-doped graphene, which is also believed to be caused by the adsorbed H atom. The small peaks near the Fermi level in Fig. 3(c) is caused by the unbonded C atoms in the vacancy, which can be explained by the narrow peak in the Fermi level in Fig. A3(b).Besides, by comparing the PDOS of these substrates during-10 - -5 eV in Figs. A2-A4, there is obvious overlap among the s orbital of the H atom and the p orbital of the C atom,and this is the reason why the interactions between H and the substrates are chemisorption rather than physisorption.
3.2. Adsorption of reactants and products on pristine graphene,N-doped graphene and vacancy graphene
After obtaining the adsorption configurations of H on different substrates,we calculated the co-adsorption of the reactants and products (i.e., CH/H, CH/2H, CH/H, CH/H,CH/2H, CH/H, CH/H, CH/2H, CH/H, CH/H,CH/2H, CH/H, CH/H, CH/2H and CH/H)involved in the Reactions (1)-(5) on pristine graphene, Ndoped graphene and vacancy graphene,respectively, and their most stable configurations are to be used as the initial states and final states during the search of TS for these reactions.After taking account of various adsorption sites and orientations of hydrocarbon molecules, the most stable adsorption configurations of the reactants and products for the CHdissociation process on H-adsorbed pristine graphene, N-doped graphene and vacancy graphene are displayed in Figs. A5-A9, respectively. And the adsorption energies are listed in Table 2. To analyze the effects of H atom on the adsorption properties of the reactants (CH, CH, CH, CHand CH) on clean and H-adsorbed pristine graphene, N-doped graphene and vacancy graphene, the comparison of the adsorption energies and adsorption distances of these configurations are listed in Tables 3 and A2, respectively. As can be seen from Tables 3 and A2, the values of all the adsorption energies and adsorption distances almost remain the same before and after the H atom is adsorbed onto these substrates,which means that the adsorbed H atom has little effect on the adsorption of these adsorbates. Besides, by comparing the adsorption energies of reactants in Tables 2 and 3,it is obvious that the values of the former ones are much larger than that of the later ones, and the differences are introduced by the chemisorption of the H atoms.
For the co-adsorption of the reactants, apart from the H atom, the radicals CHand CHare also chemisorbed on the substrates, and the molecules CH, CHand CHare physisorbed on the substrates. For the co-adsorption of the dehydrogenation products, except for the adsorption of molecules CHand CH, the adsorptions of all the products are chemisorption,and this is the reason that the adsorption energies of these configurations are much larger than that of the reactants and the H-abstraction products. However, for the co-adsorption of the H-abstraction products, except for the adsorption of molecules CH,CHand CH,the adsorption of all the products is physisorption, which is why the adsorption energies of these configurations are much smaller than that of the reactants and the dehydrogenation products. In addition, for the co-adsorption of all the reactants and products on the substrates,apart from the adsorption of molecules(i.e., CH, CH, CH, CHand H), which are physisorption, all the rest of the configurations are chemisorption. And except for the adsorption of Hon vacancy graphene that is endothermic, all the adsorption processes are exothermic.
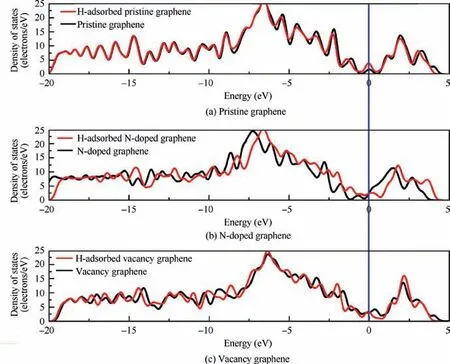
Fig. 3 Comparison of density of states before and after H atom is adsorbed on pristine graphene, N-doped graphene and vacancy graphene.
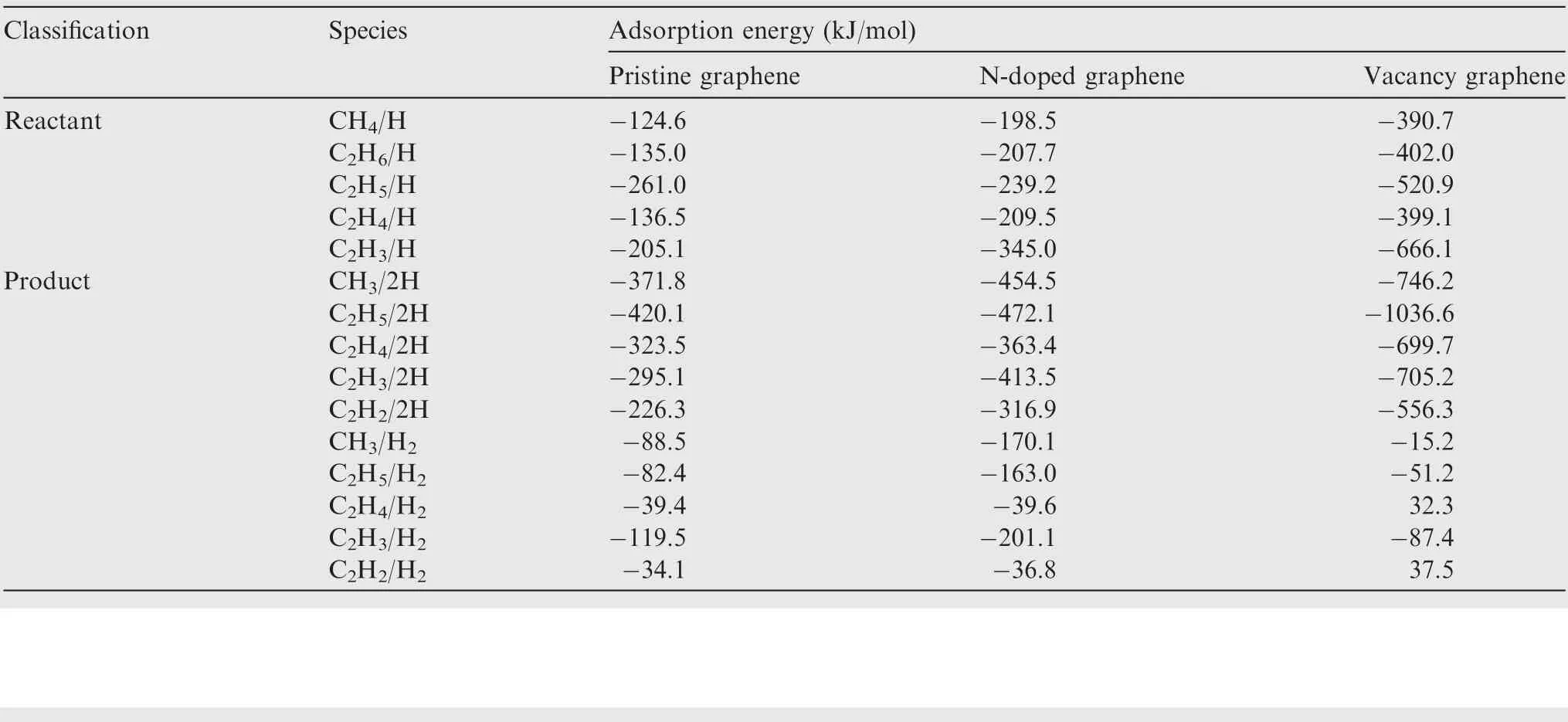
Table 2 Adsorption energies of different adsorption configurations.
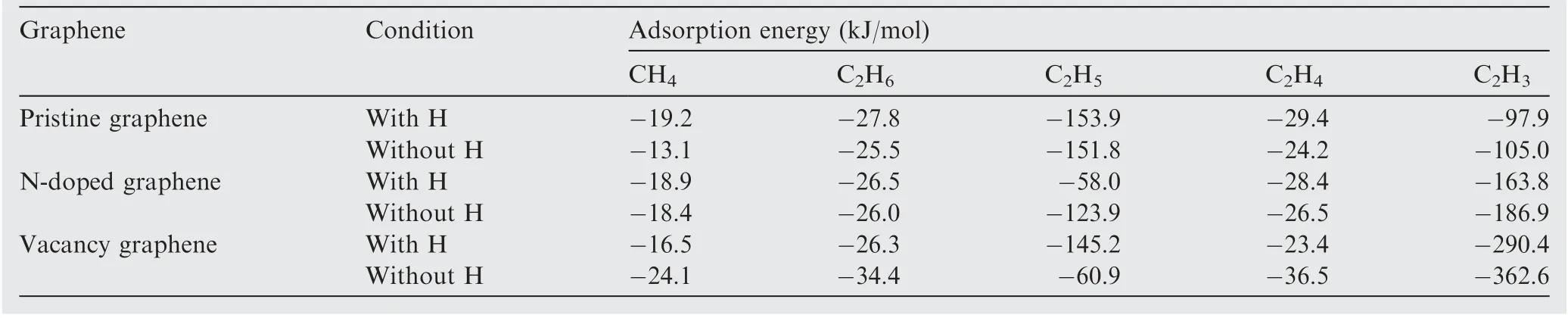
Table 3 Adsorption energies of CH4, C2H6, C2H5, C2H4 and C2H3 on pristine graphene, N-doped graphene and vacancy graphene with and without the pre-adsorbed H.
3.3.CH4 dissociation on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene
To investigate the effects of the adsorbed H atom to different graphene substrates on the dissociation mechanisms from CHto CH, the dissociation process from CHto CHon Hadsorbed pristine graphene, N-doped graphene and vacancy graphene are studied. The TS of all the reactions are calculated, and the energy profiles of each reaction are depicted in Figs. 4-8, respectively. For the convenience of comparison,the reaction pathways of the dehydrogenation reaction and H-abstraction reaction for the same reactants are displayed in the same figure,and the energy profiles of the same reaction on three different substrates are also put in the same figure.All the adsorption configurations of the reactants, TS and products are added to the energy profiles, and the green atoms in the figures represent the adsorbed H atoms. For the Habstraction reactions,there is no TS found and they are therefore thought to be barrierless reactions.
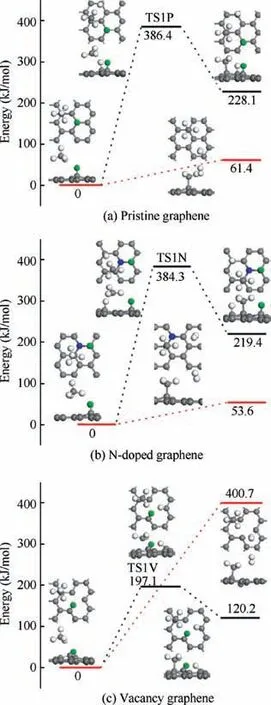
Fig. 4 Energy profiles of reaction CH4 + H →CH3 + 2H/H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.
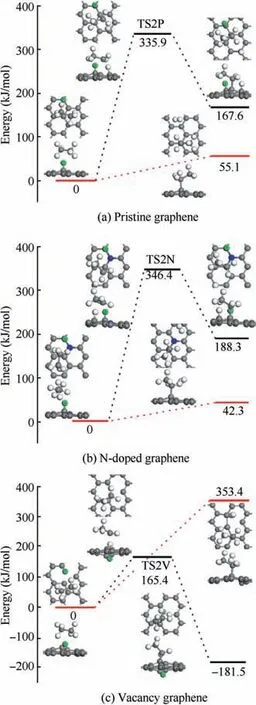
Fig.5 Energy profiles of reaction C2H6+H →C2H5+2H/H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.
3.3.1. CH→CH
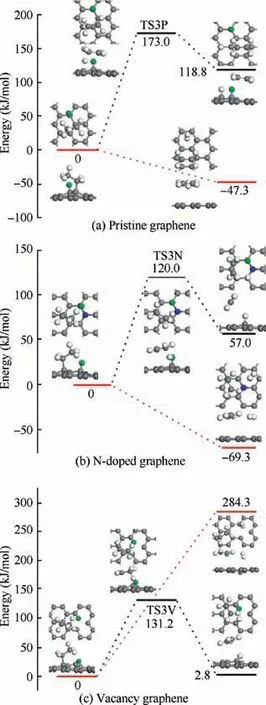
Fig.6 Energy profiles of reaction C2H5+H →C2H4+2H/H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.
For the dehydrogenation of CHon H-adsorbed pristine graphene in Fig. 4(a), the energy barrier and reaction energy are 386.4 kJ/mol and 228.1 kJ/mol, respectively, and the products are separately chemisorbed on the top sites of the substrate.The reaction energy of the barrierless H-abstraction of CHon H-adsorbed pristine graphene is only 61.4 kJ/mol. Similar to the dissociation on H-adsorbed pristine graphene,the dehydrogenation of CHon H-adsorbed N-doped graphene needs to overcome an energy barrier of 384.3 kJ/mol through TS1N with the reaction energy of 219.4 kJ/mol, and the reaction energy of the H-abstraction is only 53.6 kJ/mol. For the CHdissociation on H-adsorbed vacancy graphene, the dehydrogenation step needs to conquer the energy barrier of 197.1 kJ/mol and the reaction energy is 120.2 kJ/mol, and the H-abstraction step is performed by adsorbing energy of 400.7 kJ/mol. As indicated in Fig. 4, all the dehydrogenation reactions and H-abstraction reactions are endothermic, of which the reaction energy for the H-abstraction on the vacancy substrate is the largest, which is less favorite compared with the dehydrogenation reaction on H-adsorbed vacancy graphene. However, for the dissociation steps on both Hadsorbed pristine graphene and N-doped graphene, compared with the dehydrogenation reactions with energy barrier, the barrierless H-abstraction reactions are equipped with much lower reaction energies, resulting in that the H-abstraction is much favored than dehydrogenation on both H-adsorbed pristine graphene and N-doped graphene.
3.3.2. CH→CH
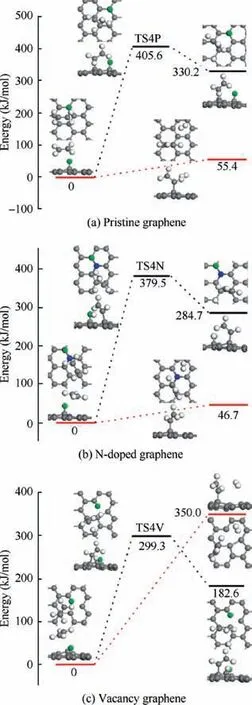
Fig.7 Energy profiles of reaction C2H4+H →C2H3+2H/H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.
For the dehydrogenation of CHon H-adsorbed pristine graphene, CHundergoes an H-elimination by overcoming the energy barrier of 335.9 kJ/mol corresponding to TS2P with the endothermic energy of 167.6 kJ/mol. And the Habstraction of CHon H-adsorbed pristine graphene is found to be endothermic with the reaction energy of 55.1 kJ/mol.The energy profiles of the reactions in Fig.5(b)look alike to that in Fig.5(a),and the activation energy and reaction energy of the dehydrogenation on H-adsorbed N-doped graphene are 346.4 kJ/mol and 188.3 kJ/mol, respectively. The reaction energy of the H-abstraction is only 42.3 kJ/mol.Different from Fig. 5(a) and (b), the two energy profiles in Fig. 5(c) are completely distinct from each other.On the one hand,the dehydrogenation on H-adsorbed vacancy graphene is accomplished by releasing 181.5 kJ/mol after overcoming the energy barrier of 165.4 kJ/mol corresponding to TS2V. On the other hand, the H-abstraction reaction is endothermic with large reaction energy of 353.4 kJ/mol. And therefore, the dehydrogenation is more favored than the H-abstraction for the CHdissociation on H-adsorbed vacancy graphene. As can be seen from Fig. 5, except for the exothermic dehydrogenation reaction on H-adsorbed vacancy graphene, all the reactions are endothermic. And due to its no barrier and low reaction energy, the H-abstraction is more favored on both Hadsorbed pristine graphene and N-doped graphene.
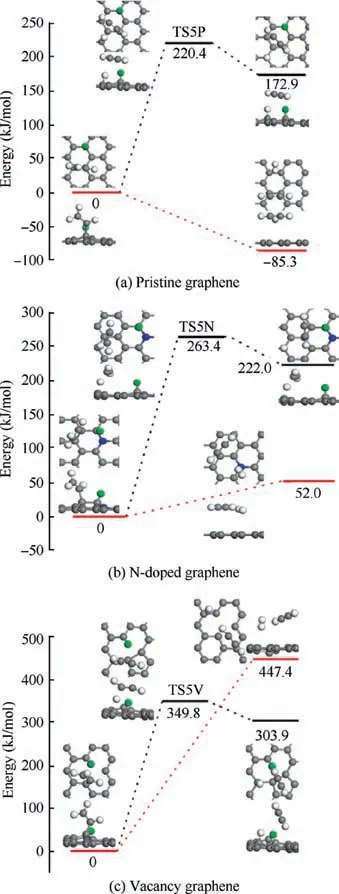
Fig.8 Energy profiles of reaction C2H3+H →C2H2+2H/H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.
3.3.3. CH→CH
As shown in Fig. 6(a), the dehydrogenation of CHon Hadsorbed pristine graphene needs to go through an energy barrier of 173.0 kJ/mol corresponding to TS3P, and the reaction energy of this endothermic process is 118.8 kJ/mol. However,for the H-abstraction of CHon H-adsorbed pristine graphene,it is exothermic and the reaction energy is-47.3 kJ/mol.For the dehydrogenation and H-abstraction of CHon Hadsorbed N-doped graphene, the energy profiles look similar to that on the H-adsorbed pristine graphene. The dehydrogenation requires the activation energy of 120.0 kJ/mol corresponding to TS3N and the reaction energy is 57.0 kJ/mol,and the energy of the products of H-abstraction reaction is 69.3 kJ/-mol lower than that of the reactants.As for the dissociation of CHon H-adsorbed vacancy graphene, the dehydrogenation step is calculated to be slightly endothermic (2.8 kJ/mol) and needs to overcome the energy barrier of 131.2 kJ/mol, and the H-abstraction step is endothermic with the reaction energy of 284.3 kJ/mol.As can be seen from Fig.6,the H-abstraction reaction is barrierless and exothermic on both H-adsorbed pristine graphene and N-doped graphene, so the Habstraction is more favored on these two substrates. Instead,the dehydrogenation is more favored on the H-adsorbed vacancy graphene because the reaction energy of the Habstraction step is too much higher than that of the dehydrogenation step.
3.3.4. CH→CH
For the dehydrogenation of CHon H-adsorbed pristine graphene,it needs to go through TS4P(shown in Fig.7(a))with a large energy barrier of 405.6 kJ/mol and the reaction energy is 330.2 kJ/mol. And the H-abstraction reaction is barrierless endothermic with the reaction energy of 55.4 kJ/mol. Similar to the energy profiles in Fig. 7(a), Fig. 7(b) shows that the CHundergoes dehydrogenation reaction on H-adsorbed N-doped graphene after conquering the energy barrier of 379.5 kJ/mol corresponding to TS4N, and the reaction energy is 284.7 kJ/mol. And the H-abstraction reaction is also endothermic with the reaction energy of 46.7 kJ/mol. For the CHdissociation on H-adsorbed vacancy graphene, the energy barrier and reaction energy of the dehydrogenation reaction are 299.3 kJ/mol and 182.6 kJ/mol, respectively, and the H-abstraction reaction is calculated to be endothermic with large reaction energy of 350.0 kJ/mol.As indicated in Fig.7,it is obvious that all these reactions are endothermic, and because of its low reaction energy and no barrier, the Habstraction is more preferred than dehydrogenation on both H-adsorbed pristine graphene and N-doped graphene. However, the reaction energy of the H-abstraction on H-adsorbed vacancy graphene is larger than that of the dehydrogenation step, which results in that the dehydrogenation is favored on this substrate.
3.3.5. CH→CH
For the CHdissociation on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene, the energy profiles of the dehydrogenation reactions and H-abstraction reactions are displayed in Fig. 8, respectively. As indicated in Fig. 8(a),the dehydrogenation reaction is calculated with an energy barrier of 220.4 kJ/mol and the reaction energy is 172.9 kJ/mol.Instead,the H-abstraction reaction on the H-adsorbed pristine graphene is exothermic with the energy of 85.3 kJ/mol.For the reactions on N-doped graphene in Fig.8(b),the energy barrier and reaction energy of the dehydrogenation reaction are 263.4 kJ/mol and 222.0 kJ/mol, respectively. And the reaction energy of the H-abstraction reaction is 52.0 kJ/mol with no TS found. As illustrated in Fig. 8(c), the reaction energies for the dehydrogenation reaction and H-abstraction reaction are 303.9 kJ/mol and 447.4 kJ/mol, and the former reaction has to overcome the energy barrier of 349.8 kJ/mol corresponding to TS5V. By comparing the energy profiles in Fig. 8, it is observed that all the reactions are endothermic except for the H-abstraction reaction in Fig. 8(a), and the H-abstraction reaction is both favored on the H-adsorbed pristine graphene and N-doped graphene for its low reaction energy. However,the H-abstraction reaction is not preferred in H-adsorbed vacancy graphene because of its high reaction energy.
3.4. CH4 dissociation mechanisms
After obtaining the energy profile of each reaction, the energy profiles of the dehydrogenation reactions and H-abstraction reactions on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene are plotted in Fig. 9(a) and (b), respectively,and the corresponding reaction energies are collected in Table 4.To make the curves consecutive,the reaction energies of 2CH→CHare calculated and added to Fig. 9. This drawing method is widely used by researchers.As can be seen from Fig. 9(a), for the dehydrogenation reactions, the total reaction energies and barrier energies on the H-adsorbed vacancy graphene are much smaller than that on the Hadsorbed pristine graphene and N-doped graphene, which indicates that the dehydrogenation reactions from CHto CHare much easier to be proceeded on the H-adsorbed vacancy graphene than the H-adsorbed pristine graphene and N-doped graphene. However, for the energy profiles of H-abstraction reactions in Fig.9(b),the total reaction energies on the H-adsorbed vacancy graphene are much larger than that on the H-adsorbed pristine graphene and N-doped graphene, which implies that the H-abstraction reactions from CHto CHare much harder to be proceeded on the Hadsorbed vacancy graphene than the other two substrates.Besides, by comparing the energy profiles of dehydrogenation reactions and H-abstraction reactions on the same substrate in Fig.9(a)and(b),it can be concluded that the dehydrogenation reactions are preferred than H-abstraction reactions on the Hadsorbed vacancy graphene, and instead, the H-abstraction reactions are more favored than dehydrogenation reactions on the H-adsorbed pristine graphene and N-doped graphene.This phenomenon can be explained by the reactivity of the vacancy site. Different from the pristine graphene and Ndoped graphene, there are three unbonded carbon atoms in the vacancy graphene, which are reactive and unstable. When the dehydrogenation reactions proceed on the vacancy site,two or three unbonded carbon atoms are connected with the products, which can help reduce the system energy and make it more stable. However, when the H-abstraction reactions are performed on the vacancy site,the H atom connected with the carbon atom of the vacancy site is pulled out of the substrate to form the Hmolecule,which can increase the number of the unbonded carbon atoms and the reactivity of the substrate by transforming the substrate into the high energy and reactive state. And this can help explain the reason why the dehydrogenation reactions from CHto CHare much easier to be proceeded than the H-abstraction reactions on the Hadsorbed vacancy graphene.
In addition, according to the previous researches,hydrogen has a clear inhibiting effect on the formation of pyrocarbon and aromatics species and the pyrocarbon deposition rates because hydrogen inhibits the homogeneous pyrolysis reactions in the gas phase and the heterogenous deposition reactions of pyrocarbon at the substrate surface.Therefore,for the consideration of the formation and growth of pyrocarbon on the substrates the dehydrogenation reactions from CHto CHon the substrates are more favored than the Habstraction reactions. Furthermore, it can be seen from Fig. 9(a) that the dehydrogenation reactions on the Hadsorbed vacancy graphene are most favored among these three substrates. And this conclusion in some way, is also in accordance with the experimental results. According to the studies,there are two kinds of carbon-hydrogen surface complexes during the dissociation of CH, a C(H) complex and a C(H) complex, which correspond to the products of the Habstraction reactions and dehydrogenation reactions, respectively. The C(H) complexes are stable until about 600 ℃,but instead, the C(H) complexes are stable up to 1400 ℃and above, especially for the C(H) complexes formed at the zigzag sites, and the reactivity of this kind of sites are similar to that of the vacancy sites. Therefore, it can be concluded that the dehydrogenation reactions on the vacancy graphene are most favored among all these reaction systems.
To further analyze the effects of the hydrogen on the dehydrogenation reactions from CHto CHon the substrates,the energy profiles of these reaction processes on the pristine graphene, N-doped graphene and vacancy graphene with and without the pre-adsorbed H atom are demonstrated in Fig. 10, respectively. The barrier energies of each reaction are collected in Table A3. As can be seen from Fig. 10, for the same substrate, the energy profile trend of each series of reactions remains the same before and after the H atom is adsorbed onto the substrate. For the reactions on the pristine graphene, N-doped graphene and vacancy graphene, the energy profiles show that there is obvious fluctuation occurred after the H atom is added to the substrates, which shows that the H atom adsorbed on the substrate has a clear effect on the CHdissociation on the pristine graphene, N-doped graphene and vacancy graphene.
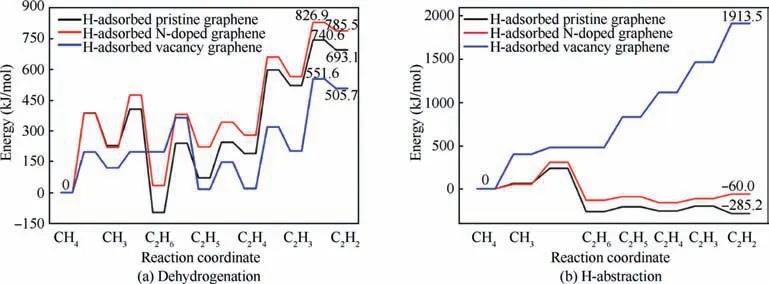
Fig. 9 Energy profiles of dehydrogenation reactions and H-abstraction reactions from CH4 to C2H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.
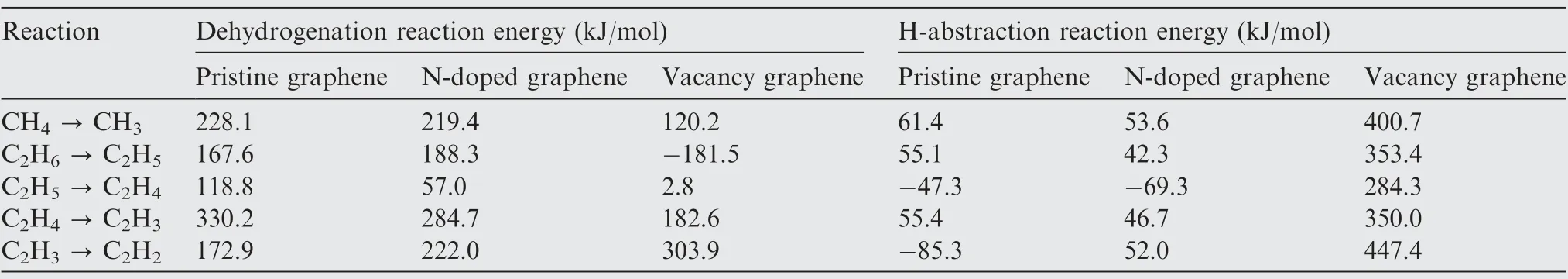
Table 4 Reaction energies involved in reactions from CH4 to C2H2 on H-adsorbed pristine graphene,N-doped graphene and vacancy graphene.

Fig. 10 Energy profiles of dehydrogenation reactions from CH4 to C2H2 on pristine graphene, N-doped graphene and vacancy graphene with and without the pre-adsorbed H.
Furthermore,the calculation results can be proved by comparing with other studies.The high reaction barriers found are in agreement with many publications,and the experimental conditionscan help confirm that these dissociation reactions can take place successfully. The chemisorption of H on graphene studied has also been proved by Balog et al.who found that the H adsorption induced a gap opening at the Fermi level. As for the conclusion that the dehydrogenation reactions are preferred than H-abstraction reactions on the H-adsorbed vacancy graphene, Li et al.has found the same phenomenon on the Cu(1 1 1) surface. And Li et al.and Losurdo et al.all have found that the adsorbed H atom has a negative effect on the formation of graphene on copper surface, which is similar to the phenomenon that hydrogen inhibits the heterogenous deposition reactions of pyrocarbon at the substrate surface. This helps confirm that the dehydrogenation reactions from CHto CHon the substrates are more favored than the H-abstraction reactions.Besides,Wang et al.demonstrated that vacancy graphene can effectively facilitate the N-H bond dissociation of ammonia and the Ndoped graphene hinders the N-H bond cleavage,which is consistent with the conclusion found that the dehydrogenation reactions are more favored on vacancy graphene than the pristine graphene and N-doped graphene.
4. Conclusions
The dissociation of CHon H-adsorbed pristine graphene, Ndoped graphene and vacancy graphene have been studied by DFT. There are two possible reactions for these three substrates,i.e.dehydrogenation reactions and H-abstraction reactions. The adsorptions of reactants are not affected by hydrogen. The dehydrogenation reactions are more favored on vacancy graphene than the other two graphene substrates,and the H-abstraction reactions are more favored on the pristine graphene and N-doped graphene than the vacancy graphene. Due to the inhibition of hydrogen on the formation and growth of pyrocarbon, the dehydrogenation reactions on the vacancy graphene are most favored among all these reactions in favor of the deposition of pyrocarbon.And after comparing the dehydrogenation reactions on these three substrates with and without the adsorbed H,the reactions on the pristine graphene, N-doped graphene and vacancy graphene are obviously affected by hydrogen.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This study is supported by the National Natural Science Foundation of China (Nos. 51821091, 51872234 and 51972271).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Appendix A.Appendix Figues and Tables to this article can be found online at https://doi.org/10.1016/j.cja.2021.12.014
 Chinese Journal of Aeronautics2022年6期
Chinese Journal of Aeronautics2022年6期
- Chinese Journal of Aeronautics的其它文章
- Experimental investigation of a gliding discharge plasma jet igniter
- Evolution of turbulent boundary layer over a three-dimensional bump
- Conceptual design and preliminary experiment of icing risk management and protection system
- Direct thrust control for multivariable turbofan engine based on affine linear parameter-varying approach
- Characteristics of reattached boundary layer in shock wave and turbulent boundary layer interaction
- Structurally coupled characteristics of rotor blade using new rigid-flexible dynamic model based on geometrically exact formulation