
PARS2 基因变异致早发性婴儿癫痫性脑病1例报告
2022-04-07 11:33王秀英李小晶朱海霞曹彬彬陈文雄伍湘玲
临床儿科杂志 2022年4期
王秀英 田 杨 石 真 侯 池 李小晶 朱海霞 曹彬彬 陈文雄 伍湘玲
广州市妇女儿童医疗中心神经内科(广东广州 510000)
早期癫痫性脑病又称早发性婴儿癫痫性脑病(early infantile-onset epileptic encephalopathy,EIEE),是一类常见的婴儿早期起病的难治性癫痫,临床表现多样,包括大田原综合征、West 综合征、早期肌阵挛癫痫脑病、婴儿恶性游走性部分性发作等[1]。2020年在线人类孟德尔遗传数据库将EIEE与发育性癫痫性脑病合并,现根据基因分型已将EIEE分为1型至93型。PARS2基因(OMIM:612036)位于1p32.3,编码脯氨酸-tRNA合成酶,后者是在蛋白质生物合成中起关键作用的酶家族,参与了RNA的表达和进程[2],基因变异可导致早期癫痫性脑病75型(OMIM:618437)。目前国内罕有PARS2基因变异病例的报道。本研究对1例EIEE患儿及其家族成员进行基因检测,发现患儿存在PARS2基因的复合杂合变异,总结其病例资料并报告如下。
1 临床资料
患儿,男,4月27日龄,因反复抽搐20余天就诊。患儿于4 月龄开始出现点头、短暂四肢弯曲强直伴哭闹发作,成串出现,每串4~6下,病初时每天发作1串,后逐渐增多至每天4~6串,多为睡醒时发作,曾就诊于当地医院病情未见好转,遂来广州市妇女儿童医疗中心就诊收住院。患儿为第2胎第2产,足月剖宫产,出生体质量3 500 g,无窒息史,围生期无异常。生后母乳喂养。父母表型均无异常,否认癫痫、发育障碍及其他疾病家族史。有一哥哥2 岁,体健。体格检查:头围38 cm,神志清醒,反应慢,可对视,能逗笑,抬头不稳,追声追物差,双足、背部、臀部见较多墨色胎记,心前区听诊无杂音,肺、肝脾、腹部查体无异常,四肢肌张力减低,各腱反射正常,病理征阴性。实验室检查:血常规、尿常规、血生化、凝血功能、电解质、免疫功能、甲状腺功能等未见异常。血酰基肉碱、氨基酸、尿代谢物筛查未见异常。脑脊液常规、生化、病原学检查未见异常。胸部X 线片、心电图、心脏超声、双耳听觉诱发电位和双眼闪光视觉诱发电位未见异常。血气中乳酸4.8 mmol/L(正常0.9~1.7 mmol/L);血乳酸/丙酮酸比值测定中乳酸 2.75 mmol/L (正常0.50~1.80 mmol/L),比值16.6。脑电图示阵发性高度失律,弥漫性中高波幅慢波背景上频繁多灶性癫痫性放电,监测到孤立性及成串痉挛发作,发作期脑电图为广泛性电压减低伴快波节律或广泛性慢波伴电压减低(图1)。头颅磁共振成像平扫+增强扫描示双侧额、顶、颞部脑外间隙增宽,脑萎缩样改变(图2)。Gesell 评分示DQ 12分,其中大运动12,精细运动12,认知16,语言10,社交12。

图1 视频脑电图表现

图2 头颅磁共振平扫及增强扫描结果
患儿表现为早发性癫痫性脑病,常规代谢物筛查未见异常,头部影像学提示脑发育不良,血乳酸水平升高,考虑可能存在遗传学病因。经父母签署知情同意书,并经医院伦理委员会批准(穗妇儿科伦批字[2019]第40101号),患儿先行染色体核型分析,结果为46,XY。然后取患儿及其父母外周血各3 mL盐析法获得基因组DNA,由于癫痫基因包和医学外显基因包的数据更新滞后,可能遗漏新的致病基因,本研究采用全外显子二代测序法进行遗传学检测,使用Illumina Nextseq 500测序平台进行测序,测序平均覆盖深度为300×,大于10×覆盖区间占100%,并对检测结果进行Sanger测序验证。应用预测软件对变异的保守性、致病性、危害性进行预测。参考美国医学遗传学和基因组学学会(ACMG)变异分类指南,对变异进行分类。结果患儿及其母亲检测到PARS2基因c.287G>A(p.Arg96His)复合杂合变异,患儿及其父亲PARS2基因c.283G>A(p.Val95Ile)复合杂合变异(图3),属常染色体隐性遗传。其中c.283 G>A 既往已多次报道,在正常人群有低频分布,其在ExAc(东亚)人群频率为0.0016,ACMG指南判断为III-意义未明;c.287 G>A变异的频率在参考人群基因数据库中未被报道,ACMG 指南判断为III-意义未明,SIFT、PP2_HDIV、ClinPred、LRT、MT、GERP 等多个计算机软件分析预测该变异为有害。

图3 患儿基因测序及家系成员验证结果
根据患儿临床及脑电图表现诊断为EIEE。给予托吡酯(TPM)6.25 mg/次,每晚1次口服,硝基安定(NZP)0.833 mg/次,每晚1次口服(体质量7.6 kg),发作控制欠佳;4天后调整托吡酯剂量为6.25 mg/次,每12小时1次,1周后硝基安定0.833 mg/次,每12小时1次,患儿发作次数减少,出院门诊随访。出院后患儿仍有发作,托吡酯逐渐加量,至第5周加量为12.5 mg/次,2次/d,硝基安定用量不变,发作明显减少,每天0~2串,每串1~3下;第6周托吡酯加量至16.6 mg/次,2次/d,患儿发作缓解。见表1。门诊随访至2021年4月,目前患儿1岁10月龄,未再抽搐发作,仍显著发育落后,抬头不稳,头围42 cm,不能独坐及站立、行走,四肢肌张力低下。Gesell评分:DQ17,其中大动作17、精细运动12、认知21、社交17、语言21。脑电图明显好转,高度失律消失,背景活动大致正常,后头部为主少量癫痫样放电。复查头颅磁共振成像较前无变化,血生化肝功能正常,血乳酸2.7 mmol/L。
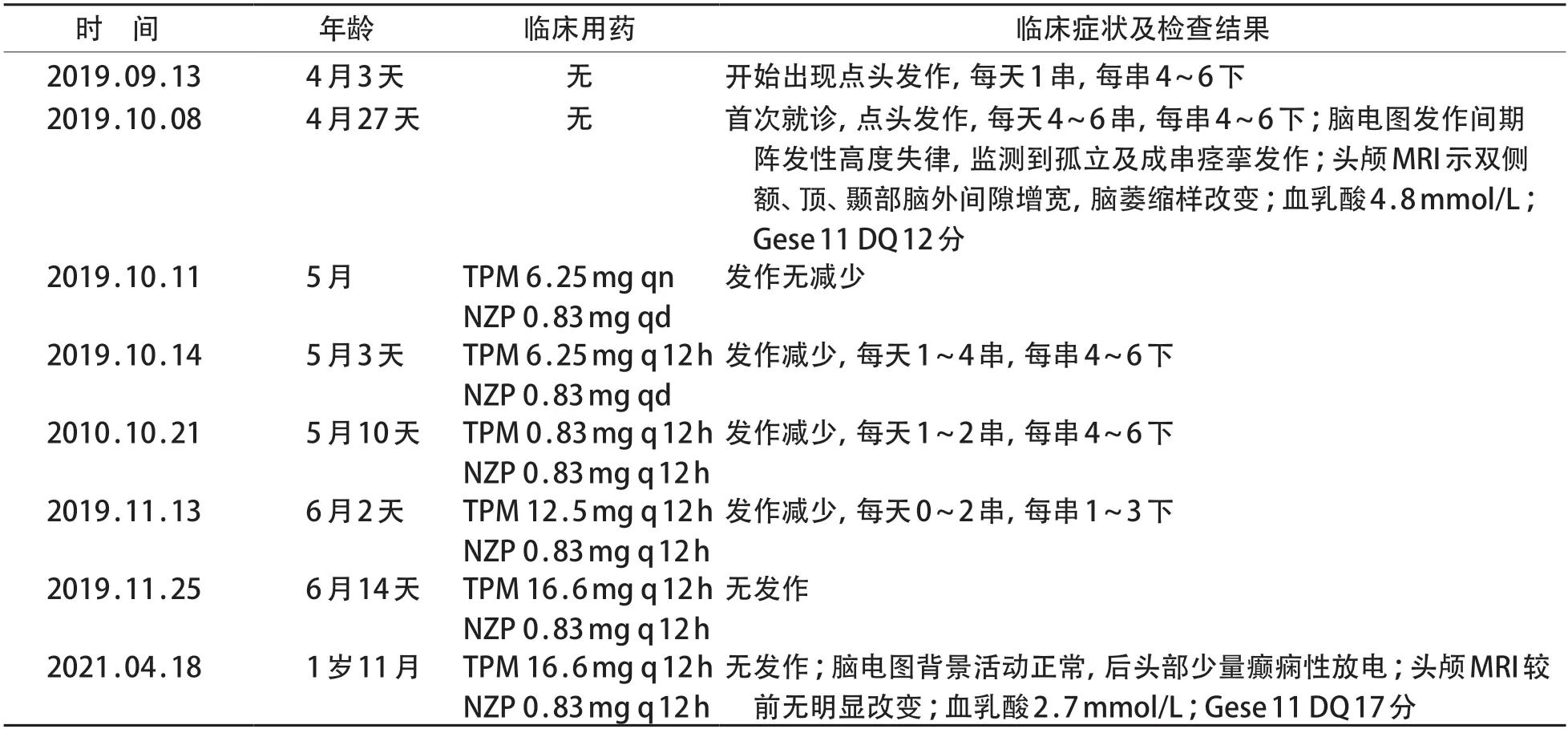
表1 患儿疾病治疗经过时间轴
2 讨论
早期癫痫性脑病的核心症状是癫痫发作并伴有发育障碍,在起病年龄、癫痫发作类型、脑电图特征、预后等方面具有高度异质性[3]。病因包括脑结构性、代谢性、遗传性等,约50%患者有可知的遗传病因,相关的变异基因有SCN1A、SCN2A、KCNQ2、STXBP1、SLC35A2等[1,4-5]。各种基因具有特定的表型谱,EIEE 的遗传学病因研究对其发病机制、基因分型、个体化治疗、预后判断及遗传咨询等具有重要意义。
PARS2基因位于1p32.3,全长1.4 kb,包含2个外显子,其编码区为第2号外显子,编码由475个氨基酸构成的二级氨基酸 tRNA 合成酶(脯氨酸-tRNA合成酶),其中47个为线粒体信号靶向的氨基酸,余428个氨基酸形成一个成熟的蛋白质。PARS2编码的脯氨酰转移酶2是氨基酰转移酶家族的II类成员,能催化脯氨酸与tRNA分子的连接[2]。人类细胞核编码的线粒体氨基酰tRNA合成酶(mt-aaRSs)由19种酶组成[6],负责催化氨基酸与其同源转移RNA (tRNA)分子的结合,从而允许每个tRNA 准确地将DNA 的遗传密码转换成蛋白质的氨基酸密码,在精确破译遗传密码方面起着重要的基础作用。除了协调核酸和蛋白质的转化,它们还可能间接影响 ATP 的产生。mt-aaRSs变异主要影响中枢神经,其他器官也参与其中[7],Sofou等[8]在2例Alpers综合征的患者中鉴定出NARS2和PARS2变异,该研究首次将PARS2基因变异与Alpers综合征联系起来。Alpers综合征是一种婴幼儿期的神经退行性疾病,其特点是大脑灰质的弥漫性变性,主要特点为精神运动进行性倒退、共济失调、难治性癫痫、卒中样发作、严重小头畸形[9]。研究发现双等位基因致病性PARS 2变异与早期婴儿癫痫性脑病(EIEE 75)相关[10],也可导致Alpers综合征[8]。
本例患儿的发病年龄、临床表现及辅助检查结果符合EIEE 诊断。基因检测发现PARS 2基因c.287G>A(p.Arg96His)复合杂合变异和c.283G>A(p.Val95Ile)复合杂合变异,常染色体隐性遗传。其中C.283 G>A(p.V 95 I)位于PARS 2高度保守的二聚体结构域,在GnomAD数据库中等位基因频率为0.00011,在ExAC中等位基因频率为0.00013,在东亚人群中为0.00161,未发现纯合子。C.283G>A(p.V 95 I) 已经在6例患儿中报道过,患儿均为早期癫痫性脑病及肌张力低下[6-7,10-12],血乳酸水平增高或正常,亚洲人种多见,可能为该基因变异的热点之一。c.287G>A(p.Arg96His)变异尚未见报道,该变异的频率在参考人群基因数据库中未被报道。按照ACMG 指南,两个杂合变异均为意义未明,但两个变异在参考人群数据库中最小等位基因分别为零和低频,多项计算机软件预测为有害,且既往已有类似变异致病的报道,本例患儿临床表型与该基因变异相关疾病高度吻合,故该复合杂合变异可能为本例患儿的致病性变异。患儿经托吡酯、硝基安定治疗后无抽搐发作,但认知和运动功能并无显著进步,这可能是PARS2基因变异所致发育障碍的特点。
以“PARS2”和“癫痫”为关键词检索PubMed数据库、中国期刊全文数据库和万方医学文献数据库自建库至2021年3月的相关文献,未检索到中文文献,检索到临床资料较齐全的国外文献6篇,排除重复报道病例,共有10例患儿。其中男4例、女6例,涉及变异位点7个,变异类型主要为错义变异,均为复合杂合变异,变异来自临床表型正常的父母,同一家庭姐弟或姐妹可同患该病[10-12]。临床特征为严重的发育落后、癫痫、小头畸形、肌张力低下、大脑结构异常,其他全身性表现包括高乳酸血症,患儿存在早期死亡的风险[8,10,12];部分患儿有视、听觉损伤及变性,以及特殊面容(前额扁平,矮鼻梁,小下颌,一字眉等);肝功能障碍只在少数患儿出现[13],这与由PLOG1变异引起的Alpers综合征不同。所有患儿均婴儿期起病,婴儿痉挛是主要发作类型,也可见肌阵挛、全面性强直-阵挛[12]、复杂部分性发作[11]。起病早期脑电图均见高度失律,治疗后高度失律可消失。头颅磁共振主要表现为进行性大脑皮层萎缩,额叶体积减少,也可表现为小脑异常信号[10],或见基底节受累[14]。部分患儿对抗癫痫药物治疗有反应,癫痫发作及脑电图明显改善。1例患儿在早期给予治疗线粒体功能障碍的抗氧化剂,结果存活至成年[13],而其未接受抗氧化剂治疗的相同表型的哥哥5岁时死于心脏衰竭,提示线粒体细胞损伤与有氧能量的减少(ATP合成减少)有关,抗氧化剂治疗或许能延长患儿的生命。目前该疾病未检素到相关的药物临床试验研究。
综上,本例患儿为新发现的PARS2基因c.287G>A(p.Arg96His)和c.283G>A(p.Val95Ile)复合杂合变异导致的早期婴儿癫痫性脑病,变异分别来自于母亲和父亲。PARS 2所致的癫痫发作多数可以药物控制,但仍有严重的认知和运动发育障碍。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国典型病例大全(2022年10期)2022-05-10
中国典型病例大全(2022年10期)2022-05-10
祝您健康(2022年2期)2022-01-14
家庭医学(2017年12期)2018-01-15
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中国医学创新(2016年33期)2017-02-28
中学生理科应试(2016年7期)2016-05-14
