
SARS-CoV-2病毒全基因组序列比对及进化分析
2022-03-26 07:53赵仁生崔艺璇许诗嘉宋鹏飞万春平
云南民族大学学报(自然科学版) 2022年2期
周 翔,赵仁生,崔艺璇,许诗嘉,宋鹏飞,温 敏,袁 燕,万春平
(1.云南民族大学 民族医药学院 民族药资源化学国家民委-教育部重点实验室,云南 昆明 650504;2.云南中医药大学 第一附属医院,云南 昆明 650021)
2019年11月新型冠状病毒在中国武汉首次被发现,在随后短短半年,新型冠状病毒给人类带来了前所未有的灾难,无数的生命的流逝,难以言计的经济损失,时至今日,疫情的影响还在延续,人们的生命,国家的安全正在受到威胁.SARS-CoV-2(Severe Acute Respiratory Syndrome Coronavirus 2)出现以来,病毒溯源和中间宿主等问题得到了民众的广泛关注,虽然只有约2%的感染者有野生动物接触史,但研究人员普遍认为SARS-CoV-2是1种来自野生动物(如蝙蝠、穿山甲等)的病毒.病毒的跨种属传播十分困难,而且往往会引起新宿主的高病死率,但SARS-CoV-2被发现以来,表现出对人类机体已经有了很好的适应性,具有传播能力强和病死率较低的特性,这与之前2003年在中国香港发现的SARS病毒和2013年中东地区爆发的MERS-CoV病毒所引起新发、突发传染病的大多数病毒有所不同[1].国际病毒分类学委员会(International Committee on Taxonomy of Viruses,即 ICTV)在2018年将冠状病毒(CoV)科分为病毒亚科和正冠状病毒亚科.病毒亚科仅包括Alphaletovirus 1个属,但正冠状病毒包含有α属、β属、γ属和δ属,总共4个属[2].目前发现,冠状病毒感染脊椎动物,主要以哺乳动物,尤其是人类和鸟类以及蝙蝠等为主.感染人类和畜类的冠状病毒多属于正冠状病毒科中α属和β属,感染禽类的多属正冠状病毒中的γ属,在猪和禽类的感染病例中也发现了部分正冠状病毒科的δ属.根据目前冠状病毒侵扰人类历史还发现在感染人群和蝙蝠的冠状病毒中,既有正冠状病毒α属也有β属,造成此次疫情的病毒源新型冠状病毒(SARS-CoV-2)正是正冠状病毒科β属[3-7].新型冠状病毒(SARS-CoV-2)为直径60~140 nm 的正链单股RNA病毒[8].其主要会感染机体上皮细胞,导致免疫功能低下的人群,尤其是老年人和小孩表现出呼吸道,消化道等疾病的临床症状.有些感染症状轻微,有些则致命.新型冠状病毒(SARS-CoV-2)主要传播途径为经呼吸道飞沫和接触传播,相对封闭条件下,高浓度气溶胶传播及粪便和尿液对环境污染造成气溶胶或接触传播[9].鉴于目前对该病毒进行的全序列分析相对较少,本论文对该病毒进行了全段位的研究分析,旨在阐述病毒的传播特点,为病毒的追根溯源提供相关依据,以及探究新型冠状病毒的易变异位点,为尽快研究出相关疫苗及可行性药物提供参考.
1 SARS-CoV-2生物信息学分析
1.1 材料筛选
从美国国立生物技术信息中心(NCBI)的GenBank核酸序列数据库中筛选出36条来自不同国家和地区的全基因序列,这些序列分别来自中国、澳大利亚、意大利、波兰、日本、俄罗斯、美国等不同国家(见表1).这些序列中最长的来自于波兰的PL_P14、PL_P13和PL_P15,全长 29 903 bp,最短的来自于俄罗斯的SCPM-0-Cdna-07,全长为 29 457 bp.另外,NCBI在参考了各国提交的序列数据后,建立了SARS-CoV-2标准全基因参考序列(NC_045512)(RefSeq),长度为 29 903 bp,即总共 29 903 个碱基,并将其作为标准序列用于与其他全基因组序列比对,从而进行SARS-CoV-2的一些特征性分析.

表1 SARS-CoV-2全基因序列及序列来源
1.2 分析方法
鉴于基因组序列的两端存在非编码区,在进行36条来自不同国家的全基因组序列比对时,采用NCBI已确定的SARS-CoV-2标准序列(NC_045512)为标准,运用其核实过的编码基因区域,即该序列的266-29 674 位碱基,总共 29 408 个碱基对,使用序列对比分析软件snapgene进行序列比较.
通过snapgene软件进行36条序列和标准序列的对比分析,找出SARS-CoV-2各基因组序列的变异位点及其在序列上的分布,以及序列上碱基对的缺失、插入和替换.从而分析推测出SARS-CoV-2的易变区域和保守区域所对应的基因序列和对应的编码蛋白质.统计所有的SARS-CoV-2序列上碱基替换数量和及其变异分布位点,找出其序列位点的变异方式,从而分析出SARS-CoV-2中碱基的易变性.
将36条来自不同国家的新型冠状病毒(SARS-CoV-2)全基因组序列进行序列对比,使用软件ClustalX1.83,采用NJ(NeighbourJoiningTrees)方法构建生物系统进化树,结合个地区分离出的病毒株变异分布,从而分析出SARS-CoV-2地区进化和病毒的流行特点.
运用软件Lasergene的程序MegAlign对36个病毒株进行同源性分析,将参考序列NC_045512作为参考对象,对应与其进行同源性比对分析.
2 分析结果
2.1 自不同地区的SARS-CoV-2全基因组序列变异数量分布
统计分析来自不同国家地区的36条SARS-CoV-2全基因组序列中266-29 674 位碱基,总计 29 408 bp.发现大部分基因序列都存在变异,且变异的类型主要以碱基替换为主.36条全基因组序列经比对分析,共存在128处变异位点,其中包括2个来自俄罗斯的病毒株在64个位点上的碱基缺失,33个病毒株在64个位点上发生的碱基替换.统计发现,36条全基因组序列,共计发生碱基缺失128次,碱基替换次数达190次,总计发生变异发生的次数达到318次,平均每条序列变异8.8次,变异率在0.3‰(318/29 408/36).36个病毒株的变异数量分布见表2和图1.
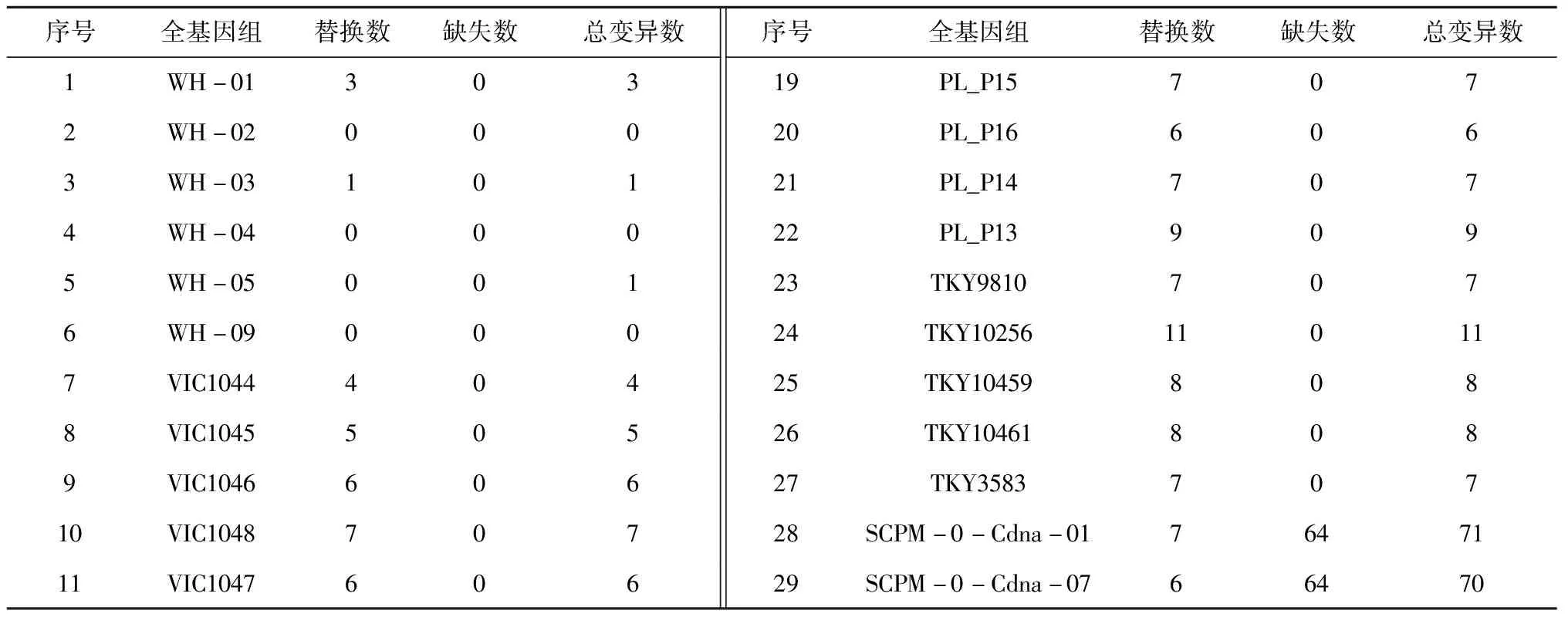
表2 SARS-CoV-2全基因组变异数量分布
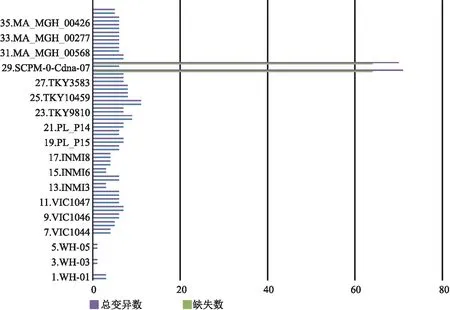
图1 36个SARS-CoV-2全基因组序列变异数量比较
其中没有发生变异的是来自中国武汉的3个病毒株,分别为WH-02、WH-04和WH-09;变异数量较少(≤3)的病毒株有5个,它们是来自中国武汉WH-01、WH-03、WH-05和来自印度的INMI3、INMI6;变异数量较多(≥10)的病毒株有3个,这3个序列是来自俄罗斯的SCPM-0-Cdna-01、SCPM-0-Cdna-07和来自日本的TKY10256.
从变异数量的分布来看,来自俄罗斯的2个病毒株SCPM-0-Cdna-01在29544-29608位点和SCPM-0-Cdna-07在29457~29521位点(相当于SARS-CoV-2参考序列的29610~29674位点)碱基对缺失较多.由于缺失的问题也导致了这2个病毒株的序列编码蛋白和其他序列有所区别.
2.2 变异位点在SARS-CoV-2全基因组序列上的分布
对36条来自不同国家和地区的SARS-CoV-2病毒株全基因组序列的全长 29 408 bp 的碱基替换位点数量和碱基缺失位点数量以及碱基插入数量进行了统计比对(见表3),其分布趋势(见图2).结合了NCBI网站上提供的各编码蛋白质类别及分布位点(见图3),对各编码蛋白质基因区域的变异位点及类型进行了统计(见表4).分析对比SARS-CoV-2各蛋白编码区间类别(见表4和图2),可以观察到,在全基因组序列中,碱基替换发生在3k~4k、14k~15k、23k~24k和28k~29k区间相对比较多,这些区间主要是ORF1ab蛋白区域和S蛋白及N蛋白编码区.碱基缺失主要发生在29k~30k区域,主要是ORF10蛋白区域.在28k~30k区域,即ORF10蛋白区域和N蛋白区域内碱基的缺失和碱基替换的位点数量达到最高.
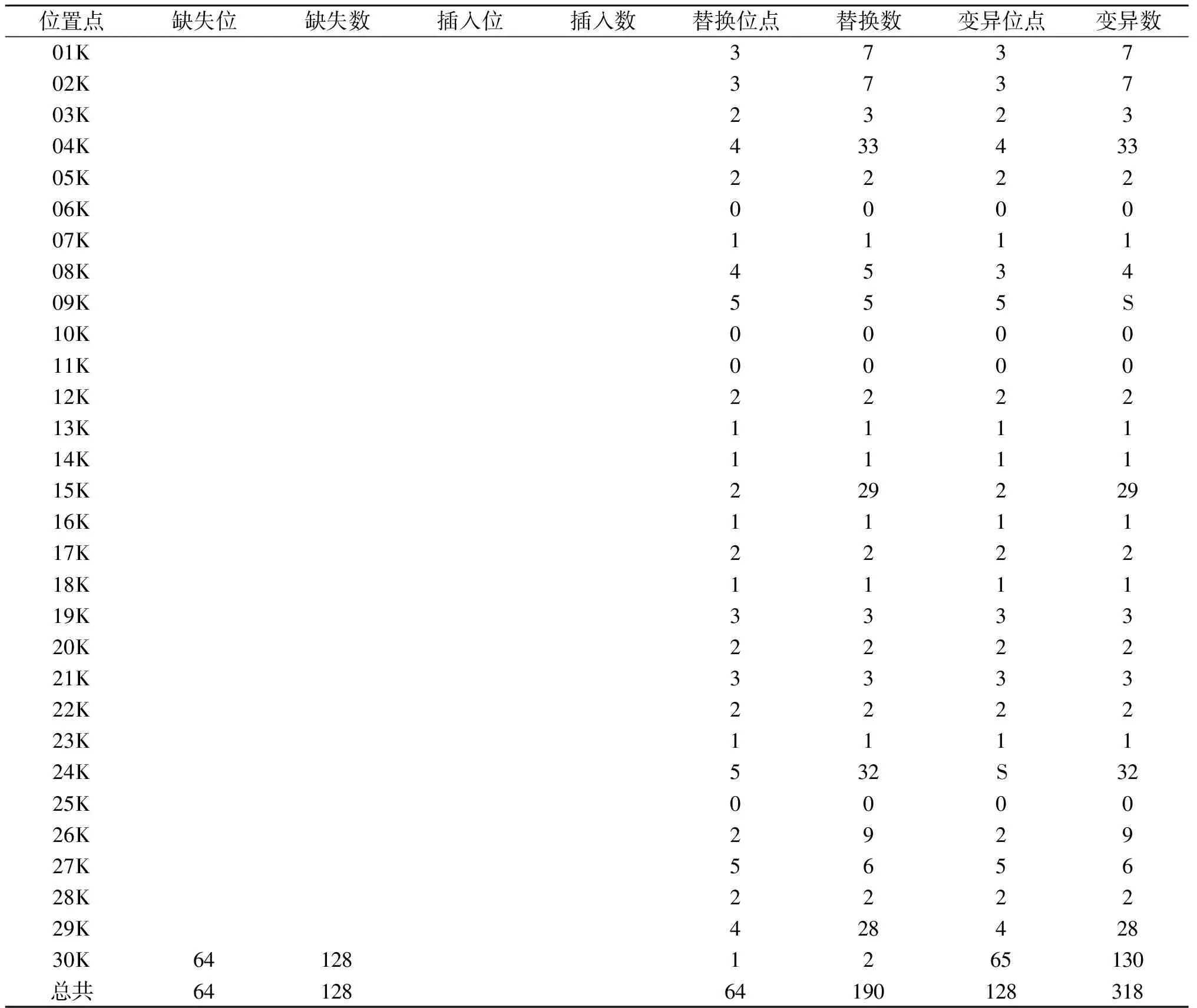
表3 36条SARS-CoV-2全基因组上碱基变异位点和数量分布
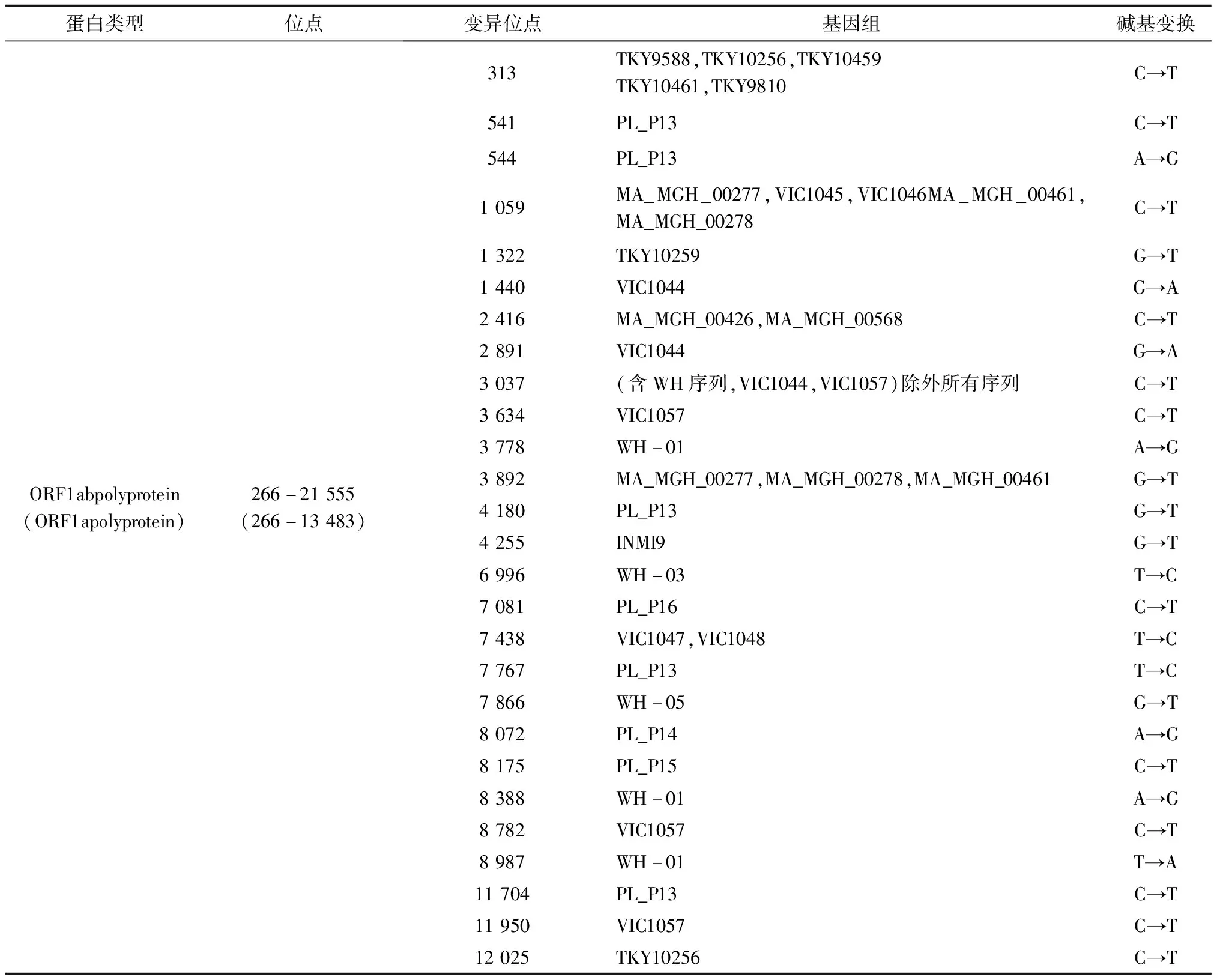
表4 SARS-CoV-2全基因组序列上蛋白编码区域及变异位点分布
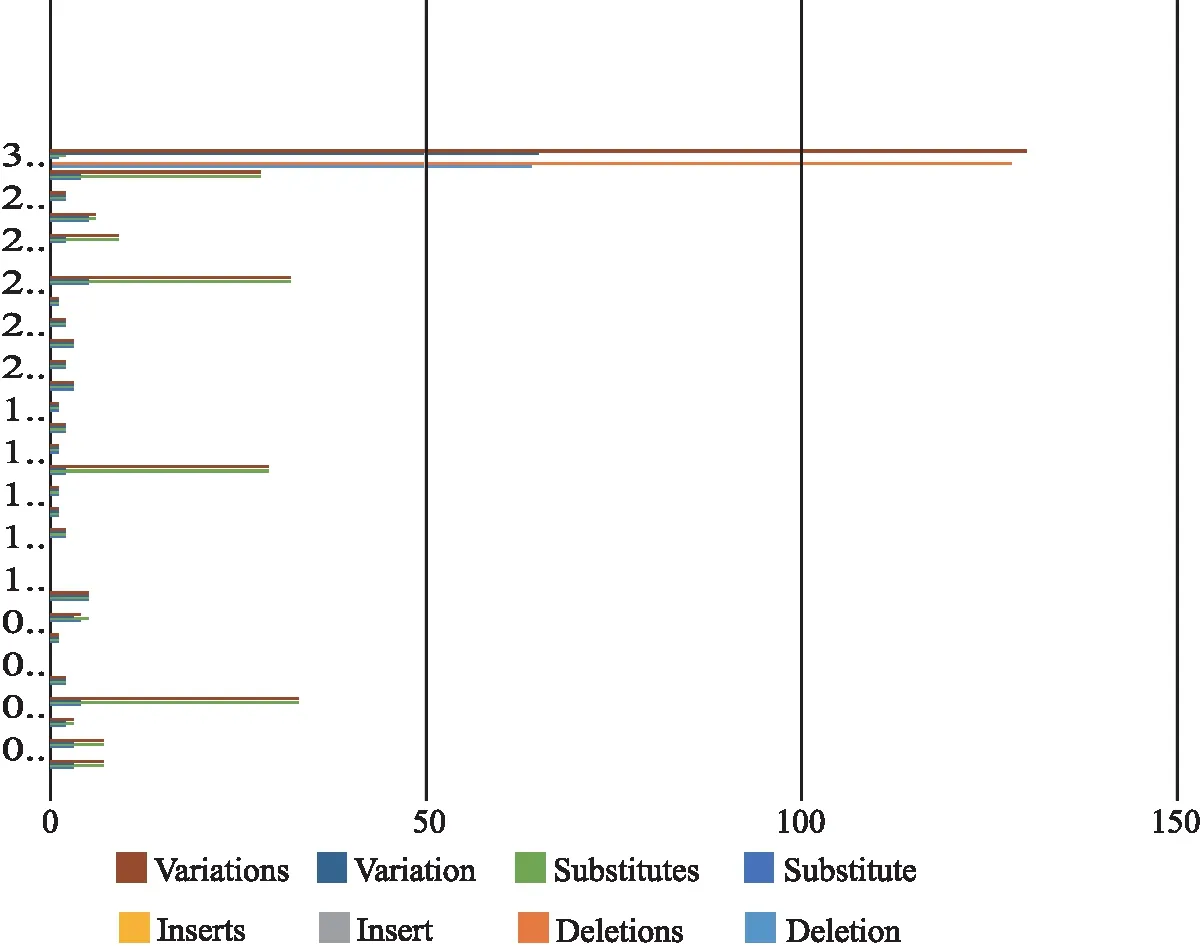
图2 36条SARS-CoV-2全基因组序列上变异位点及数量对比
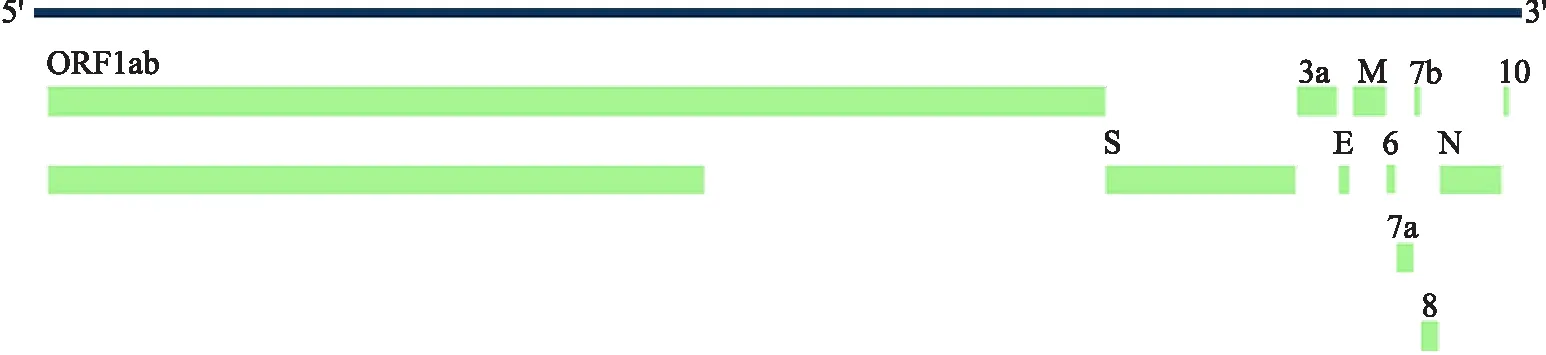
图3 各蛋白在全基因组序列中的编码区域分布
在SARS-CoV-2全基因序列中,碱基变异多数集中在序列的后半区域,也就是发生在3′一端的23k后面的区域,即23k~30k之间.后半段的序列编码包括S蛋白、M蛋白、E蛋白和N蛋白等重要的结构蛋白.除去ORF10蛋白区域的碱基缺失,变异的集中区域在S蛋白区域(21 563~25 384)和N蛋白区域(28 273~29 533).在变异的位点和数量上来看,S蛋白编码区域变异位点8个,总变异数量达35个,N蛋白编码区域变异位点3个,变异数量达29个,N蛋白重复变异率较高.除此之外,E蛋白和M蛋白的变异位点及变异数量相对较低,值得注意的是ORF6蛋白区域和ORF7b蛋白区域没有发生变异.
SARS-CoV-2全基因序列中,变异的类型以碱基替换和碱基缺失为主.碱基的缺失发生在来自俄罗斯的2个病毒株(SCPM-0-Cdna-07和SCPM-0-Cdna-01),其缺失区域为ORF10蛋白编码区域,几乎缺失了整个编码区域.碱基的替换发生的位置较多,S蛋白区域的碱基替换以A→G类型为主,N蛋白区域的碱基替换以G→A为主,位点23k前段区域的碱基替换以C→T为主.
2.3 SARS-CoV-2生物进化分析
使用Cluastal X软件对36个SARS-CoV-2病毒株的全基因序列进行多序列比对(Do Complete Alignment),再利用其进化树功能绘制NJ进化树图(Neighbour Joining Trees),绘制出来后再运用MEGA7.0进行进化树调整及分析,见图4.
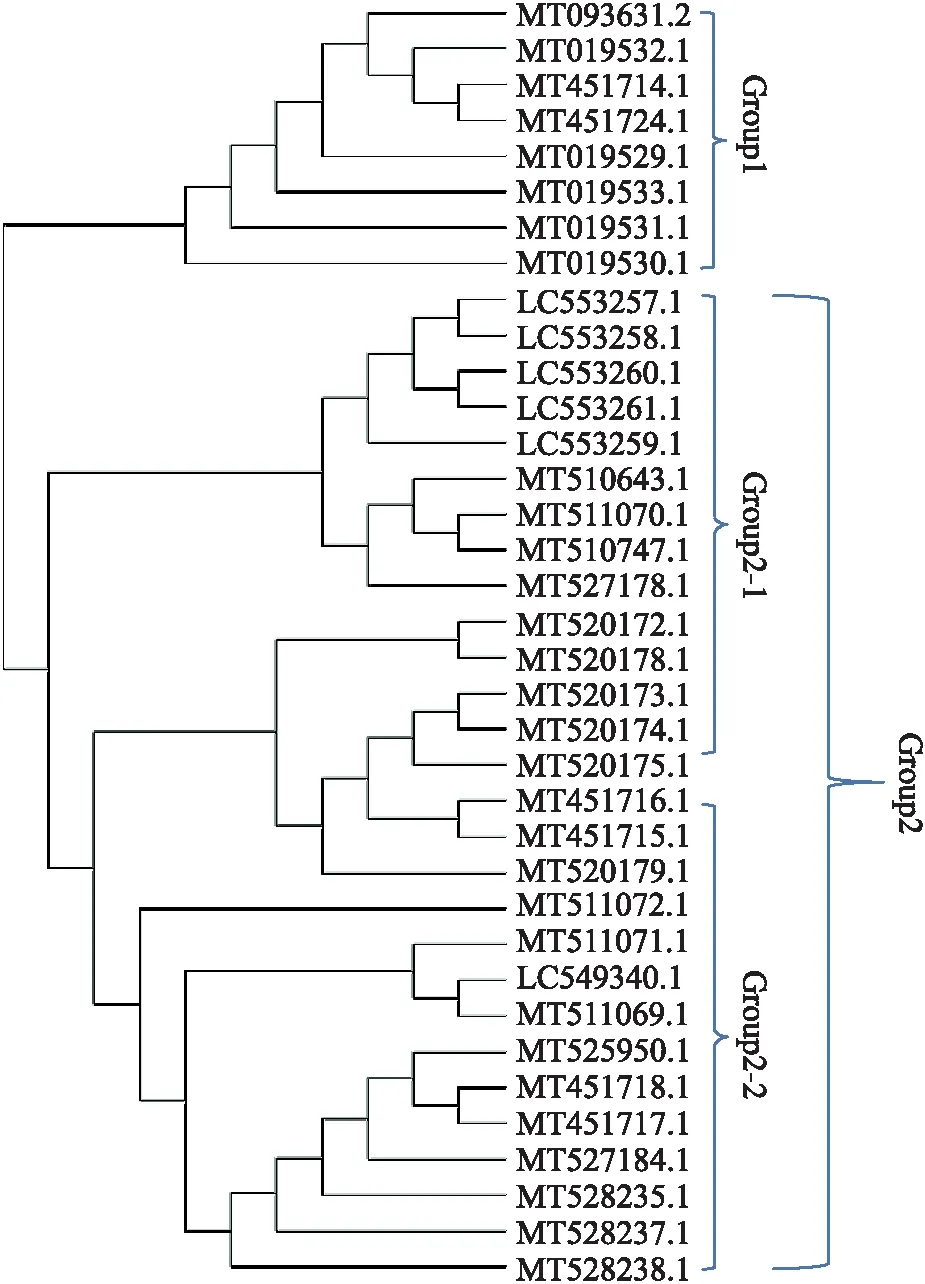
图4 基于SARS-CoV-2全基因组的遗传进化分析
通过调整之后得出的进化树可以将36个序列分成2群组,第2群组里面又可以分成2小群组.从图中可以看出,第1群组(Group 1)8个病毒株大部分都来自SARS-CoV-2病毒首次被发现的地方,即中国武汉,而且序列提交的时间都在2—3月.另外还有2条来自澳大利亚的病毒株,与源自中国武汉的病毒株亲缘性较高,序列提交的时间在5—6月,可以认为病毒株在此时间段内并未发生太大的变化.第2群组(Group 2)涵括有28个病毒株,而这28个病毒株又可以分为2小组,即Group 2-1和Group 2-2.Group 2-1该群组包括有9个病毒株分别来自日本、波兰、俄罗斯和意大利,SARS-CoV-2病毒株的提交时间都在5月20日~6月10日之间,其中5条来自日本,另一分支上的4条序列来自其他3个国家,亲缘性较高,可以看出病毒株极有可能在这个时间段内伴随着国际交通路线四处传播.另外1个群组(Group 2-2),包含了剩下的19个SARS-CoV-2病毒株,其中6个来自美国的病毒株和2个来自澳大利亚的病毒株占据了1条分支,提交时间在5—6月中旬,此群组亲缘性较高.剩下的一条进化分支上包含了3个来自波兰的病毒株,1个来自日本的病毒株,2个来自澳大利亚的病毒株,以及5个来自意大利的病毒株,提交时间都在5—6月中旬.综合进化树可以看出,流行于美国等国家的SARS-CoV-2病毒株与中国武汉传播的SARS-CoV-2病毒株有着一定的区别,其亲缘性并不是很高.
2.4 SARS-CoV-2同源性分析
通过Lasergene软件的Megalign小程序对36株选自不同地区国家的病毒株进行同源性分析,其基因组同源性分析结果如下表5.
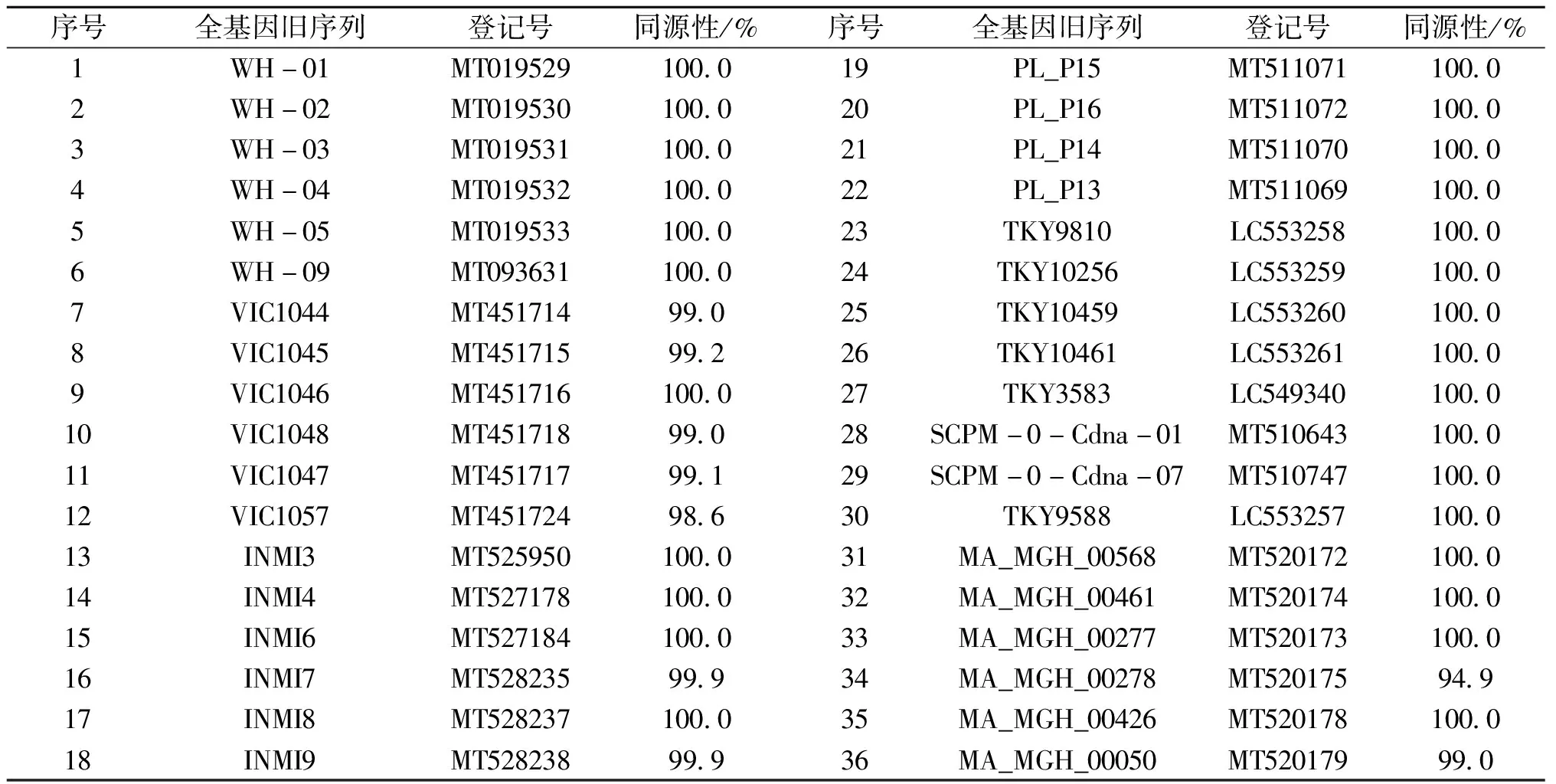
表4 SARS-CoV-2参考病毒株NC_045512与其他病毒株的同源性
以病毒株NC_045512为参考序列,与其他病毒株进行比对,得出同源性差异.从表中可以看出参考序列NC_045512与其他病毒株同源性是相当高的,基本都在99%以上.除了来自美国的序列MA_MGH_00278与参考序列的同源性只有94.9%,以及来自澳大利亚的序列VIC1057与参考序列同源性为98.6%.可以说,在同源性上,所有病毒株与参考病毒株并没有多大差异,具有高度同源性.
3 讨论与分析
综合来看,筛选的36条SARS-CoV-2全基因组序列大部分都存在变异的现象,除开两端非编码区,全基因组序列总的变异率在0.300‰(318/29 408/36),ORF1ab蛋白编码区的变异率在0.142‰(109/21 289/36),S蛋白编码区域的变异率在0.254‰(35/3 821/36),N蛋白编码区的变异率在0.639‰(29/1 260/36),M蛋白编码区的变异率在0.208‰(5/668/36),E蛋白编码区的变异率在0.122‰(1/227/36).S蛋白是糖蛋白的1种,由两个重要的结构域构成,S1亚基纤突蛋白受体结合域和S2亚基糖蛋白,具有多个抗原表位,能刺激机体从而使机体产生抗体,该蛋白与机体宿主细胞ACE2受体的亲和力是SARS病毒的10-20倍,因而SARS-CoV-2病毒能够更快的与人体细胞结合使机体出现病症,以及在人群中更快的传播[10].M蛋白是病毒跨膜蛋白,即糖基化的基质蛋白的1种,在病毒的组装和出芽中起作用[11].N蛋白是结构蛋白的1种,主要负责SARS-CoV-2病毒的组装[12].E蛋白控制SARS-CoV-2病毒的组装,是其包膜的组成成分,E蛋白如果变异缺失,可以导致SARS-CoV-2病毒株丧失使机体致病的能力[13].结合上述各蛋白的变异率来看,E蛋白的变异率最低,N蛋白的变异率最高,S蛋白的变异率其次,可以看出病毒在传播过程中,为了适应环境从而感染不同地区和国家的人群,在结构上发生进行了相应的变异,以达到和机体宿主细胞ACE2受体的结合.根据其致病力一直居高不下,可以看出其E蛋白编码区的高度稳定保守.SARS-CoV-2病毒基因组在ORF1ab蛋白编码区具有高度变异性,虽然基本都是沉默变异.在SARS-CoV-2全基因组中还存在另外一处存在高度变异性的区域,即ORF8蛋白编码区,变异率为0.076‰(1/365/36),由于ORF8蛋白存在多态性,其变异会导致2个变异体ORF8-L和ORF8-S,从而导致其蛋白质的结构异常[14].除了上述蛋白编码区域外,36条SARS-CoV-2病毒全基因组序列值得注意的其他区域还有ORF6蛋白编码区,高度保守没有发生任何变异.其次,在36条SARS-CoV-2全基因组序列中,来自俄罗斯的两条基因组序列尤其值得注意,两条全基因组序列全部缺少ORF10蛋白编码区,是整个编码蛋白区的缺失,可以猜想,在俄罗斯传播的SARS-CoV-2病毒株大多不含有ORF10蛋白.从病毒株在缺少ORF10蛋白的情况下,仍能感染人群来看,ORF10蛋白在病毒株的重组过程中是可以舍弃的.当然也存在另外一种可能,由于其缺失的主要是3′端的基因片段,那就是与测序准确率和基因组拼接等有关.
虽然现在还未确定SARS-CoV-2病毒株的来源,但根据已有的研究数据及进展来看,该病毒不太可能来源于已知家畜、家禽以及犬猫等宠物冠状病毒[15].在疫情早些时候,Zhou等发现SARS-CoV-2基因组序列与蝙蝠身上携带的冠状病毒(Bat CoV RaTG13)的序列一致性达到了96.3%[16].同时,中科院等院校通过分析Global Initiative on Sharing All Influenza Data(GISAID)数据库的93个SARS-CoV-2基因组数据发现:病毒是外源流入该市场内,并引起大面积传播与蔓延[17].结合36株来源于不同国家地区的SARS-CoV-2病毒株的全基因组序列分析及通过软件分析的出的生物进化树,可以确定在美国等地区流行的SARS-CoV-2病毒与中国地区流行的SARS-CoV-2病毒的亲缘性并不是很高.运用Lasergene软件的Megalign程序对所有序列进行了同源性比较,可以得出的结论就是流行于美国等地区与流行于中国地区的SARS-CoV-2病毒是同一种病毒,都是SARS-CoV-2病毒.综上所述,从美国国立生物技术信息中心(NCBI)的GenBank核酸序列数据库中筛选出36条来自不同国家和地区的全基因组序列都是属于SARS-CoV-2病毒基因组序列,再根据进化树上的进化分支及节点来看,流行于美国的SARS-CoV-2病毒与中国流行的SARS-CoV-2病毒来源于同一祖先,但是处在不同的分支,其进化历程方向是不同的,因此,传闻SARS-CoV-2病毒是“以中国为源头,向全世界传播”这种说法是没有依据的.
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
科学之谜(2021年2期)2021-04-25
科学导报(2020年54期)2020-09-09
支部建设(2020年15期)2020-07-08
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
电脑知识与技术(2018年19期)2018-11-01
百科知识(2015年18期)2015-09-10
