
放射性治疗药物的非临床安全性评价策略探讨
2022-03-03 13:35杜春燕张艺哲邢红艳汪溪洁
中国药理学与毒理学杂志 2022年12期
杜春燕,赵 琪,张艺哲,邢红艳,李 华,汪溪洁
(1.中国医药工业研究总院,上海 201203;2.上海益诺思生物技术股份有限公司,上海 201203)
放射性药物(radiopharmaceuticals)是含放射性核素标记的用于机体内医学诊断或治疗的化合物或生物制剂。按用途分为放射性诊断药物和放射性治疗药物,按核素类型分为α、β和γ核素标记物。放射性药物由放射性核素、靶向配体[1-2]及两者之间的连接臂[3]3部分组成。随着核医学的发展,放射性药物需求不断增加,利用以同位素示踪技术为基础的放射性药物为探针,靶向机体内各生物大分子,并结合单光子断层扫描仪(single photon emission computed tomography,SPECT)或正电子断层扫描仪(positron emission computed tomography,PET)进行生物分子功能显像已广泛应用于研究体内靶分子的生理及病理等基本过程[3-4]。据美国国立卫生研究院(National Institutes of Health,NIH)网站ClinicalTrials.gov统计,截至2021年8月,放射性药物临床研究近1000项。随着SPECT和PET/PET-CT的普及,以99mTc,18F和68Ga为放射性核素的放射性诊断药物得到迅速发展,18F-脱氧葡萄糖(18-fluorine flurode oxyglucose,18F-FDG)是目前临床应用最广泛的肿瘤代谢显像剂。
相较于放射性诊断药物,放射性治疗药物临床应用较少,从最初的131I[NaI]用于甲状腺肿瘤和甲亢的治疗[5],至今,美国食品药品监督管理局(Food and Drug Administration,FDA)共批准上市了多菲戈(二氯化镭223,radium-223 dichloride,Xofigo)[6]、131I-间位碘代苄胍(iobenguane131I,Azedra)[7]、镥氧奥曲肽(lutetium Lu-177 dotatate,Lutathera)[8-9]和177Lu-维特四氧西坦(lutetium Lu-177 vipivotide tetraxetan,Pluvicto)4种放射性治疗药物。α粒子靶向治疗和肽受体放射性核素疗法等广泛应用于神经内分泌瘤靶向治疗领域[9-10]。随着国际上放射性治疗药物的不断研发,其非临床安全性评价也因其特殊结构面临巨大挑战。本综述结合国内外指导原则,通过分析放射性治疗药物的主要毒性风险及部分已上市放射性治疗药物的非临床安全性评价方法,探讨其非临床安全性评价策略。
1 放射性治疗药物的毒性风险
由于放射性治疗药物结构特殊且复杂,即使标记同一放射性核素的药物也可能因为靶向配体不同导致药物毒性存在差异,因此我们从放射性核素、靶向配体和放射性治疗药物3方面探讨放射性治疗药物可能存在的毒性风险。
1.1 放射性核素
目前,常用的放射性核素包括131I,89Sr,90Y,177Lu和223Ra等。放射性核素通过电离辐射生物效应靶向治疗肿瘤,但发射α粒子的核素射程短、能量积聚快、寿命短(221Fr,t1/2=4.8 min;217At,t1/2=32.3 ms;213Bi,t1/2=45.6 min)[11],导致其以极高浓度聚积在肿瘤细胞或肿瘤细胞簇附近,引起细胞毒性。放射性核素经历100~200 keV的反冲能[12]后脱离靶向剂,游离的放射性核素可能是剂量限制性毒性的来源[13]。如在一项研究225Ac标记的抗大鼠人类表皮生长因子受体2单克隆抗体试验中观察到的长期肾毒性,便是由于放射性核素225Ac反冲于肾且在肾保留率高导致的[14]。
1.2 配体
放射性治疗药物的配体多为小分子、抗体、多肽和蛋白。与放射性核素诱导的毒性相比,配体的毒性通常比较轻微,但一些放射性治疗药物由于配体过量,也会产生药理毒理相关不良反应。如来昔决南钐[153Sm]〔samarium(153Sm)lexidronam,153Sm-EDTMP〕中未标记的EDTMP过多时会络合体内微量元素并排泄至体外,产生不良反应。
1.3 放射性治疗药物
当放射性核素与配体通过连接臂结合成整体后,可能会产生以下毒性:①细胞毒性。在一项131I-[Tyr3]奥曲肽〔131I-[Tyr3]octreotide,131I-TOC〕治疗Graves眼病试验中[15],对比观察131I,131I-TOC以及单独奥曲肽对成纤维细胞的毒性作用。结果发现仅131I-TOC造成细胞形态改变,如体积增大、颗粒增多和膜结构崩解,且随放射剂量增加,细胞存活率也显著降低(P<0.05)。②肾毒性。Miederer等[11]在一项评估225Ac-偶联人源化抗 CD33抗体HuM195的毒性试验中,当给药剂量为215和189 kBq·kg-1、累积剂量为377 kBq·kg-1时,食蟹猴肌酐水平升高、贫血恶化,导致肾功能衰竭;且组织病理学显示,肾组织损伤可能与间充质纤维化有关。③迟发放射毒性。一项多发性骨髓瘤的Ⅰ/Ⅱ期放射性药物治疗试验显示[16],约30%(30/80)患者出现了2~4级肾功能损伤,其中7人发展为严重血栓性微血管病变,需要进行肾透析(7例中5例已死亡);在一项放射性同位素90Y标记的奥曲肽(90Y-DOTAD-Phe1-Tyr3-octreotide,90Y-DOTATOC)治疗晚期神经内分泌瘤的Ⅱ期临床试验中,36例中5例出现慢性肾功能衰竭[17],其中3例人由血栓性微血管病变引起。
2 放射性治疗药物的非临床安全性评价
放射性药物的非临床安全性评价需先遵循《支持药物进行临床试验和上市的非临床安全性研究指导原则》〔Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals,ICH M3(R2)〕。针对放射性治疗药物,美国FDA颁布了《放射性治疗药物晚期辐射毒性的工业非临床评价指南》(Guidance for Industry Nonclinical Evaluation of Late Radiation Toxicity of Therapeutic Radiopharmaceuticals)《放射性肿瘤治疗药物:非临床研究和标签推荐工业指南》(Oncology Therapeutic Radiopharmaceuticals:Nonclinical Studies and Labeling Recommendations Guidance for Industry);欧洲药品管理局(European Medicines Agency,EMA)颁布了《放射性药物非临床要求指南》(Guideline on the Non-clinical Requirements for Radiopharmaceuticals);目前国内尚未颁布放射性治疗药物相关指导原则。结合上述国外放射性治疗药物相关指导原则,遵循科学合理、具体问题具体分析的原则,总结、探讨放射性治疗药物非临床安全性评价中需考虑的要点。
2.1 一般研究设计
为满足非临床安全性评价研究,至少需要完成药动学-毒动学试验、安全药理学试验、单次给药毒性试验和重复给药毒性试验[18],针对特殊药物还需考虑开展遗传毒性试验、生殖与发育毒性试验或致癌性试验等。放射性治疗药物安全性评价中应关注人体各器官耐受剂量的安全阈值以及人体试验的伦理限制,非临床安全性评价还需考虑按照药物非临床研究质量管理规范和“动物福利法”的要求开展放射性治疗药物的迟发放射毒性研究,以降低其迟发放射毒性风险[19-20]。动物种属的选择和剂量设计是上述毒理学试验顺利开展的关键。
2.1.1 种属选择
用于放射性治疗药物非临床安全性评价的动物应与人体放射剂量、生物分布和药动学等具有相似性。研究表明,大鼠和犬在经历放射性核素电离辐射后发生晚期放射性肾病和肺纤维化[21-22];兔和犬可见放射性心肌纤维化[23-24]。若无合适动物模型应考虑替代试验。种属选择也因配体不同而有差异。若配体为大分子(如单克隆抗体),应参考《生物制品的临床前安全性评价》〔Preclinical Safety Evaluation of Biotechnology-derived Pharmaceuticals,ICH S6(R1)〕,该指导原则建议种属选择需考虑目标序列同源性、相对靶点结合的亲和力、受体/配体占位以及动力学等因素,当无相关物种存在时应考虑使用表达人受体的相关转基因动物或使用同源蛋白;若配体为小分子,《支持药物进行临床试验和上市的非临床安全性研究指导原则》〔Guidance on nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals,ICH M3(R2)〕建议急性毒性试验和重复给药毒性试验采用2种动物种属(啮齿类和非啮齿类)。美国FDA《放射性肿瘤治疗药物:非临床研究和标签推荐工业指南》指出在研究放射性肿瘤治疗药物生物分布、放射剂量以及配体诱导的毒性评价时,如有科学依据单一动物种属即可。
2.1.2 剂量水平
放射性治疗药物的非临床安全性评价通常设置多个剂量组来体现剂量反应特征,迟发放射毒性研究建议设计4个剂量水平,拟设剂量应体现短期内的放射毒性效应,高剂量应至少是人体最大拟用剂量或关键脏器放射耐受剂量的2倍,此剂量下应观察到放射性治疗药物毒性的发展及转归,此外还应考虑不具有放射性的受试物的总剂量,除非其剂量很低(如微克剂量)。
2.2 药动学-毒动学研究
药动学-毒动学研究提供放射性治疗药物的吸收、分布、代谢和排泄等信息。生物分布是放射性治疗药物药动学-毒动学研究重点。FDA建议生物分布研究考虑以下几点。①种属选择:如有科学依据,单一物种种属足够,在选择物种时,应考虑所有相关信息包括药理学活性和药动学特征等。②数量选择:大动物(猴)的生物分布和剂量测定通常采用影像学方法,少量动物(3雌和3雄)足以评估活性水平随时间的分布;对于小动物(大鼠和小鼠),放射自显影时每个时间点需要足够的动物数量。③性别选择:雄性和雌性动物都应被纳入特定器官摄取放射性的研究,除非具有性别特异性[25]。④数据采集时间:应考虑衰变产物及半衰期,即有足够的采样时间(如5倍有效半衰期)来生成时间-放射性活度值曲线,数据采集的持续时间可根据需要进行调整(当有效半衰期较长或时间活性曲线为多指数时,可能需要更多的采样时间点和增加动物数量)。⑤靶器官:一般包括肾上腺、骨骼、骨髓、大脑、小肠壁、大肠壁、胃、心、肾、肝、肺、肌肉、卵巢、胰腺、脾、睾丸、胸腺、甲状腺、膀胱和子宫[26];如有充分理由,评估的器官数量可减少,但至少应包括骨髓、肝和肾等高暴露器官。此外,应基于药物特点补充观察器官,如黑色素结合物应观察眼和皮肤。⑥其他方面:考虑临床生物分布和剂量学研究,由于混合剂量中放射性和非放射性部分的比例会影响生物分布,在动物研究中的使用比例应与患者使用的比例相当。
对于配体为化学药的受试物,应采用不具有放射性的受试物开展体外代谢和药物相关作用研究。对于长半衰期放射性治疗药物,可用放射性治疗药物开展体外稳定性和体内代谢研究。收集尿、粪便和胆汁检测放射性,分析放射性排泄过程。
2.3 非临床安全性评价
非临床安全性评价至少应完成安全药理学研究和一般毒性研究(单次给药毒性试验和重复给药毒性试验)。针对特殊药物,应根据需要考虑开展遗传毒性、生殖与发育毒性、致癌性及迟发毒性研究。
2.3.1 安全药理学
安全药理学试验参考《人用药品安全药理学试验指导原则》(Safety Pharmacology Studies for Human Pharmaceuticals,ICH S7A)和《人用药品延迟心室复极化(QT间期延长)潜在作用的非临床评价指导原则》〔The Non-clinical Evaluation of the Potential for Delayed Ventricular Repolarization(QT interval prolongation)by Human Pharmaceuticals,ICH S7B〕。放射性治疗药物的安全药理学评价一般采用不具有放射性的受试物开展试验。安全药理学研究试验终点可纳入毒理学或动物体内生物分布研究的设计中,安全性评估仅需详细观察啮齿动物和非啮齿动物给药后的临床反应以及测量非啮齿动物的心电图。此外,生物分布研究结果可进一步发现放射性治疗药物对体内重要器官系统(心血管、呼吸和中枢神经系统)的潜在不良影响,如放射性治疗药物分布到中枢神经系统可预示中枢神经系统中辐射诱导的血管异常、脱髓鞘和坏死引起的解剖学和功能性神经缺陷的可能性[27]。除动物试验外,体外KCNH2基因(human ether-a-go related gene,hERG)编码的钾通道实验也是安全药理学评价中重要的一部分,检测指标半数抑制浓度(half-inhibitory concentration,IC50)评估放射性治疗药物对离子电流的影响。
2.3.2 一般毒理学
放射性治疗药物的一般毒理学研究包括支持首次人体临床试验(first in human clinical trials,FIH)的一般毒理学研究和支持上市的一般毒理学研究。
美国FDA指导原则建议支持FIH的一般毒理学研究大体分2种情况。①若放射性治疗药物是纯放射性核素(不含配体),毒性仅来自于放射性核素的衰变,在进行FIH之前可不单独开展毒理学研究,附加安全终点(临床特征、体重、血液学、血清化学等)的动物体内生物分布研究结果可用于确定短期辐射相关毒性。②若既有放射性核素又有配体,核素诱导的毒性评价可不单独开展一般毒理学研究,动物生物分布研究结果以及已有的器官特异性辐射诱导毒性文章足以确定辐射毒性;配体诱导的毒性评价在启动FIH之前,先对不具有放射性的受试物进行一般毒理学研究,通常选择2个种属(啮齿类和非啮齿类);若配体无生物活性,临床为微剂量,考虑一般配体的毒性比放射性毒性弱,因此开展一种相关动物的一般毒性试验即可。放射性治疗药物支持FIH的给药剂量应注明放射性给药剂量(即给药活度)和药物的质量剂量。给药频率遵循《抗肿瘤药物非临床评价指导原则》(Nonclinical Evaluation for Anticancer Pharmaceuticals,ICH S9)且覆盖临床给药频率。一般毒理学研究通常按临床给药方式进行设计,以毒动学、血液学、临床化学、大体解剖和组织病理学等作为评估终点并重点观察辐射敏感的检测指标。根据放射性治疗药物的特点考虑增加检测频率、延长观察周期等,如若临床需多次或重复给药,则应采用临床拟用途径开展重复给药毒性试验。放射性治疗药物在体内存留时间较长时,毒理学试验周期也应相应延长。需要注意的是,如不具有放射性的受试物的毒理研究结果与放射性治疗药物之间具有明显不同的药动学特征,则建议采用具有放射性的放射性治疗药物开展重复给药毒性试验。
一般来说,支持FIH研究的非临床数据和临床Ⅰ期数据足以支持进入Ⅱ期,但由于放射性不仅引起急性损伤(照射后几天至几周内损伤)还可引起迟发性损伤(辐射后几月甚至几年后发生),因此在开展Ⅲ期临床或上市之前应进行长期毒性研究,即支持上市的一般毒理学研究,并与上市申请一并提交。应采用2种动物种属(啮齿类和非啮齿类)按临床拟用途径开展重复给药毒性试验,给药周期可参考ICH M3(R2)的要求,若放射性治疗药物在体内存留时间较长时,毒理学试验的观察周期相应延长。对于大多数治疗晚期癌症患者的放射性药物,为期3个月的非临床研究足以支持上市。对于配体诱导的毒性评价,若患者给药次数有限(2或3次)、配体仅用于递送目的、给药剂量小(微克范围)、给药频率低(每隔4~8周)、不具有放射性的受试物无明显生物活性且半衰期短等,则可不单独开展不具有放射性的受试物慢性毒性研究,美国FDA建议若要进行为期3个月的慢性毒性研究,一般认为单一种属即可;若患者预期寿命较长,可能出现迟发放射毒性时,需要进行迟发放射毒性评价,试验应考虑辐射分布(动物生物分布和人类放射剂量学研究)和描述迟发放射影响的文献或者进行动物试验。
目前美国FDA的指导原则主要适用于放射性肿瘤治疗药物,已上市的药物适应证也均为肿瘤。开展放射性治疗药物的非临床安全性评价时,还应根据各国药品监管机构的要求,结合核素和配体的特性,遵循具体问题具体分析的原则,科学合理地进行研究设计,充分评估放射性治疗药物的安全性。
2.3.3 遗传毒性
放射性核素具有造成生物体遗传物质损伤的潜力,因此需要考虑放射性治疗药物的遗传毒性风险。对于适用于晚期癌症的抗肿瘤药物,美国FDA建议在药物开发或批准期间不需要进行放射性药物或不具有放射性的受试物的遗传毒性研究,因为α,β和γ射线辐射会造成DNA损伤并具有遗传毒性风险,非临床遗传毒性研究可豁免但应在药物产品标签中列出相应的风险,如用于治疗去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)且无内脏转移的放射性治疗药物Xofigo,非临床安全性评价研究中未对223Ra进行遗传毒理学研究。但作为一种发射α粒子的放射性治疗剂,高能量的α射线导致相邻细胞双链DNA断裂,足以表征Xofigo具有遗传毒性,只需在药物标签上列出遗传毒性风险即可[28]。若放射性治疗药物的非放射性部分为新结构化合物,应对非放射性部分进行遗传毒性的评估,建议参考ICH M3(R2)和《人用药物遗传毒性试验和结果分析指导原则》〔Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use,ICH S2(R1)〕进行基础的非临床遗传毒性研究,如评估药物致突变和致分裂潜能的体外试验——细菌回复突变试验(Ames试验、哺乳动物染色体畸变试验)在Ⅰ期之前进行;体内试验(大小鼠微核试验)应在Ⅱ期之前进行,原因是非放射性部分的潜在毒性可能与放射性核素的遗传毒性效应具有相加或协同作用[29]。如在Azedra的非临床遗传毒性试验中,采用鼠伤寒沙门菌株进行体外细菌回复突变试验,采用人外周血淋巴细胞进行体外染色体畸变分析试验,体内进行了小鼠骨髓微核试验,试验结果均为阴性,未见明显毒性。
2.3.4 生殖毒性
放射性治疗药物所携带的放射性核素可产生辐射,对胎儿或生殖能力有潜在损伤。对于携带放射性核素的抗肿瘤药物,美国FDA建议通常不需要进行非临床生殖毒性研究,但需要在产品标签上注明缺乏动物生殖毒性数据和对胎儿或生殖能力潜在伤害未知的声明[30]。如目前已批准上市的放射性治疗药物——Xofigo,Lutathera和Azedra在非临床安全性评价中均未进行任何生殖毒性研究。对于非抗肿瘤药物,通常也不需要单独对非放射性部分进行生殖毒性研究,除非放射性核素的辐射较低且药物治疗时间过长,才考虑进行非临床生殖毒性研究,具体试验设计建议参考ICH M3(R2)和《人用药物生殖与发育毒性检测》〔Detection of reproductive and Developmental Toxicity for Human Pharmaceuticals,ICH S5(R3)〕指导原则。
2.3.5 致癌性
美国FDA指导原则指出在药物开发或批准期间,不需要进行放射性治疗药物或其不具有放射性的受试物的致癌性研究。原因是α,β和γ射线辐射会造成DNA的损伤,辐射是致癌的主要诱导因素,致癌性属是剂量考虑范围之内。若放射性治疗药物的非放射性部分为新结构化合物,具体试验设计建议参考药物致癌性试验相关指导原则(ICH S1AS1C)要求进行致癌性评估。
2.3.6 迟发放射毒性
临床上,放射性药物治疗后至少几个月或几年后才能观察到迟发放射毒性。如大鼠放射性肾炎的潜伏期为3~7个月不等[31],犬在立体定向放射治疗10个月后显示Ⅰ级中枢神经系统毒性[32]。因此,如果放射性治疗药物有迟发放射毒性风险,美国FDA《放射性治疗药物晚期辐射毒性的工业非临床评价指南》指导原则建议给药后至少1年内监测动物的迟发辐射毒性。非临床研究设计应模拟预期临床试验的设计,包括动物和人之间相似的放射活性剂量、给药频率、给药间隔、相对组织转化率、相对生物分布和药动学等。迟发辐射毒性的监测指标与常规的单次给药毒性试验和重复给药毒性试验相似,包括临床观察、进食量、体重、眼科检查、血液学、血清化学、尿液分析和病理(大体解剖、器官重量和组织病理)等。
迟发放射毒性研究应至少包括4个剂量水平,以确定未观察到有害作用水平(no observed adverse effect level,NOAEL)以及与该剂量相关的轻度至重度迟发辐射毒性。若药物存在非放射性核素,研究还需包括不具有放射性的受试物剂量对照组以区分特定的放射效应和与非放射性核素相关的药理作用(除非不具有放射性的受试物的剂量很低,如微克剂量)。选择的最高剂量应产生急性放射毒性,该剂量至少是人体最大拟用剂量或关键器官辐射耐受剂量的2倍,在临床试验中被确定为可能的剂量限制因素;应观察到毒性的发展及转归,在临床检测指标的采样检测方面,间隔不应短于3个月。按照具体问题具体分析的原则设计,每组动物的剂量应确保有足够数量的动物存活,以便在研究完成时进行适当的分析。
3 已上市放射性治疗药物非临床安全性研究
自2013年以来,FDA相继批准了Xofigo,Lutathera,Azedra和Pluvicto 4种放射性治疗药物,这4种药物所完成的非临床安全性评价试验有一定的参考价值,也为后续放射性治疗药物的药理毒理学试验的开展提供了方向。
3.1 Xofigo
Xofigo由拜耳(Bayer)公司研发,于2013年5月15日获美国FDA批准,用于治疗晚期转移型CRPC[33]。2020年8月,中国国家药品监督管理局批准其氯化镭[223Ra]注射液用于治疗伴症状性骨转移且无已知内脏转移的CRPC。该药物所完成的非临床毒性试验[34]见表1。
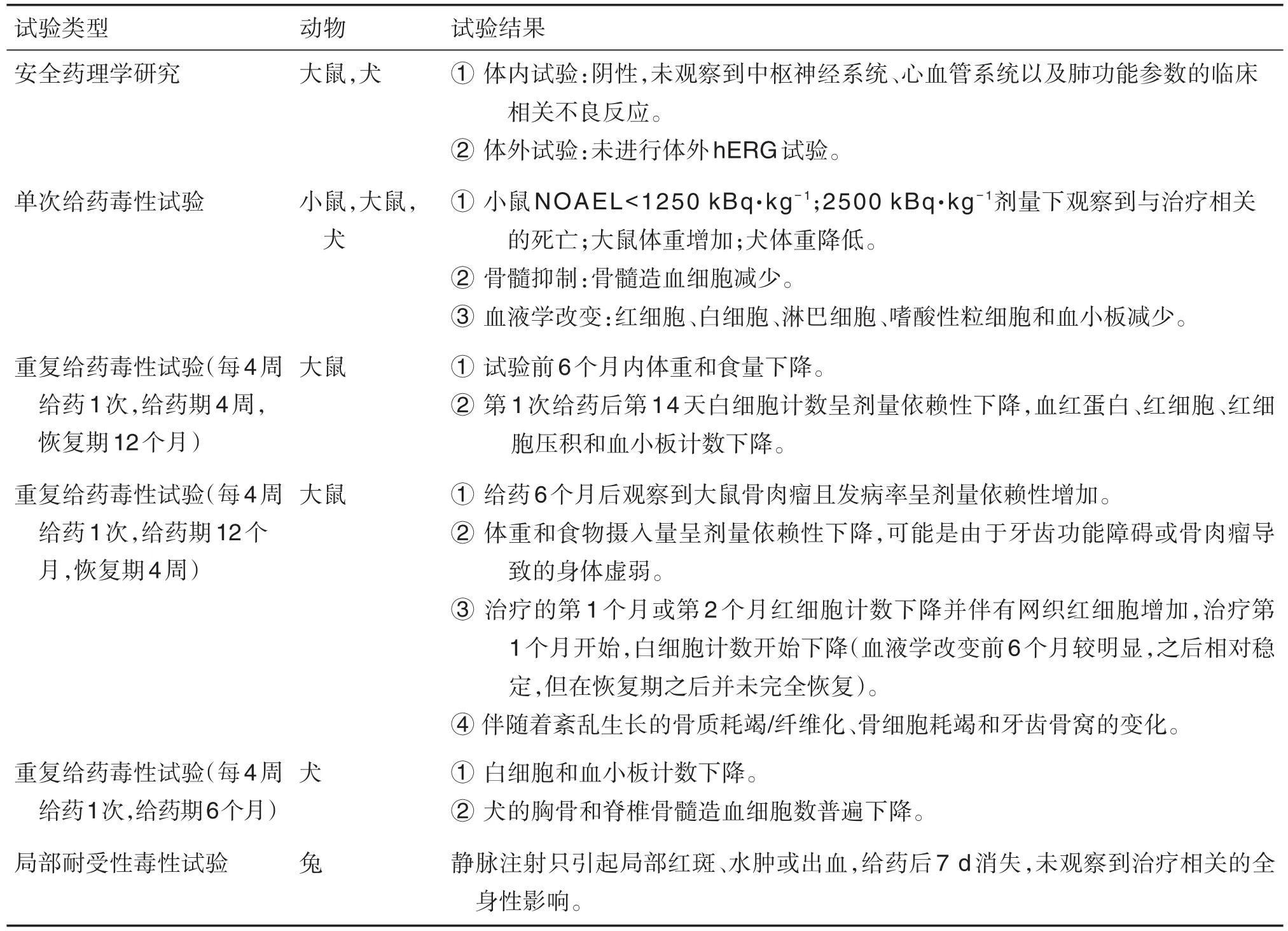
表1 Xofigo非临床毒性试验及结果[34]
该公司在临床前对Xofigo进行了单次给药毒性试验和重复给药毒性试验,目的在于发现Xofigo潜在毒性。重复给药毒性试验观察指标包括死亡率、临床体征、体重、进食量、血液学与凝血、骨髓检查、血清化学、尿检、眼检、心电图、大体病理、组织病理、器官重量检查和毒动学等。重复给药毒性试验中大鼠和犬均出现血液学毒性相关症状,如白细胞计数、血小板计数相比于对照组均下降;红细胞数下降但网织红细胞数目增多(表明红细胞再生);恶性骨肿瘤(骨肉瘤)等治疗相关变化。
3.2 Azedra
Azedra是由Progenics公司开发的用于治疗罕见肾上腺肿瘤(手术无法切除且肿瘤已扩散)的药物[35],由放射性核素131I和肾上腺素能神经元阻断剂碘苄胍(iobenguane,MIBG)通过共价键链接而成。所完成的非临床毒性试验[36]见表2。
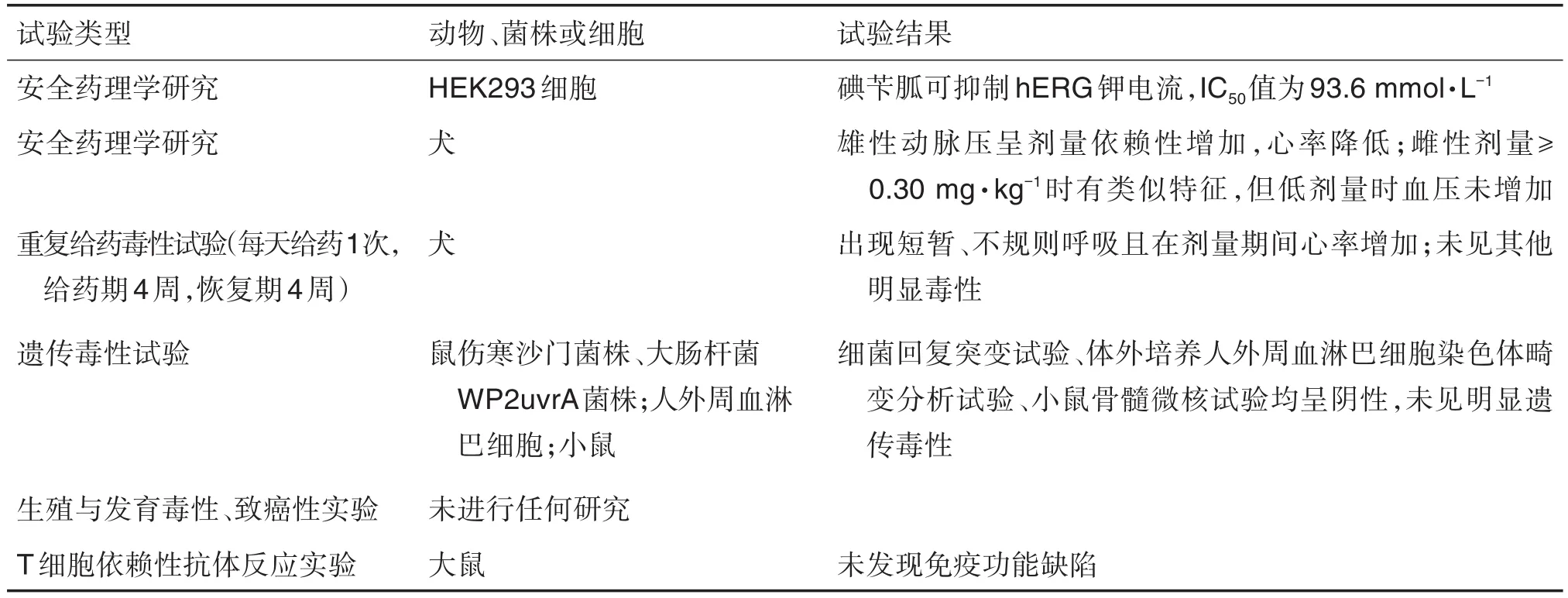
表2 Azedra完成的非临床毒性试验及结果[36]
Progenics公司在非临床安全性评价中主要对131I-MIBG进行了Ames试验、体外染色体畸变试验和微核试验,以检测药物潜在的遗传毒性。非临床安全性评价中采用不具有放射性的受试物127I-MIBG开展试验,它是合成131I-MIBG的交换标记方法的产物[37]。相较于127I-MIBG,高比活度的131I-MIBG抗肿瘤活性高,但在去甲肾上腺素转运蛋白高表达器官中放射性暴露水平增加,相关毒性也增加[38]。如在一项犬心血管安全药理研究中,未标记MIBG剂量≥0.3 mg·kg-1时,出现明显动脉压升高和反射性心动过缓;127I-MIBG 1.085 mg·kg-1对犬hERG介导的QT间期无任何影响。重复给药毒性试验观察指标包括死亡率、心电图、器官重量、组织病理和毒动学等。127I-MIBG遗传毒性试验在剂量范围内显示阴性,无明显毒性。131I-MIBG本身属于放射性产品,存在生殖和发育毒性,非临床安全性评价试验中未进行生殖毒性评价。
3.3 Lutathera
Lutathera是由诺华子公司法国Advanced Accelerator Applications(AAA)公司开发的用于治疗生长抑素受体阳性胃肠胰腺神经内分泌肿瘤的放射性药物。Lutathera是一种177Lu标记的生长抑素类似物,通过与含生长激素抑制素受体的细胞结合而发挥作用,是全球首个获批的肽受体放射性核素疗法药物[39]。所完成非临床毒性试验[40]见表3。
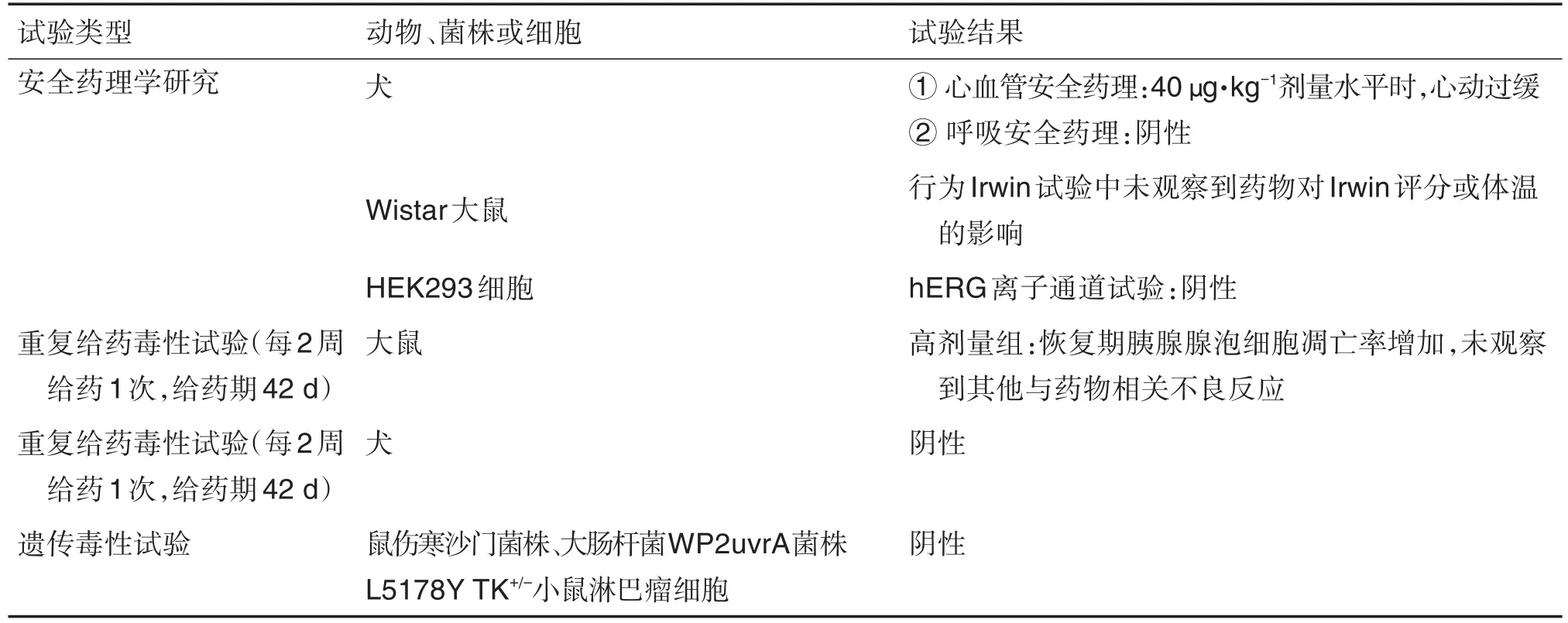
表3 Lutathera完成的非临床毒性试验及结果[40]
177Lu-DOTA0-Tyr3-Octreotate的非临床安全性评价试验多数采用不具有放射性的受试物175Lu-DOTA0-TYR3-Octreotate进行,177Lu和175Lu唯一的区别是放射性标记不同,核素175Lu适用于评估预期临床产品非放射性配体介导的毒性。175Lu-DOTA0-Tyr3-Octreotate在Ames细菌致突变性试验中无致突变性,在体外对小鼠淋巴瘤细胞无致突变性。177Lu-DOTA0-Tyr3Octreotate药物本身是放射性产品,存在遗传毒性,与ICH S9指南一致[41];非临床安全性评价试验中也未进行生殖毒理研究。
3.4 Pluvicto
Pluvicto是由诺华公司研发的用于治疗已接受基于紫杉烷的化疗和雄性激素受体信号通路抑制剂治疗的前列腺特异性膜抗原(prostate specific membrance antigen,PSMA)阳性转移型转移型CRPC患者,于2022年3月23日获美国FDA批准上市。Pluvicto是首款美国FDA批准的用于治疗此类mCRPC患者的靶向放射配体疗法。所完成非临床毒性试验[42]见表4。
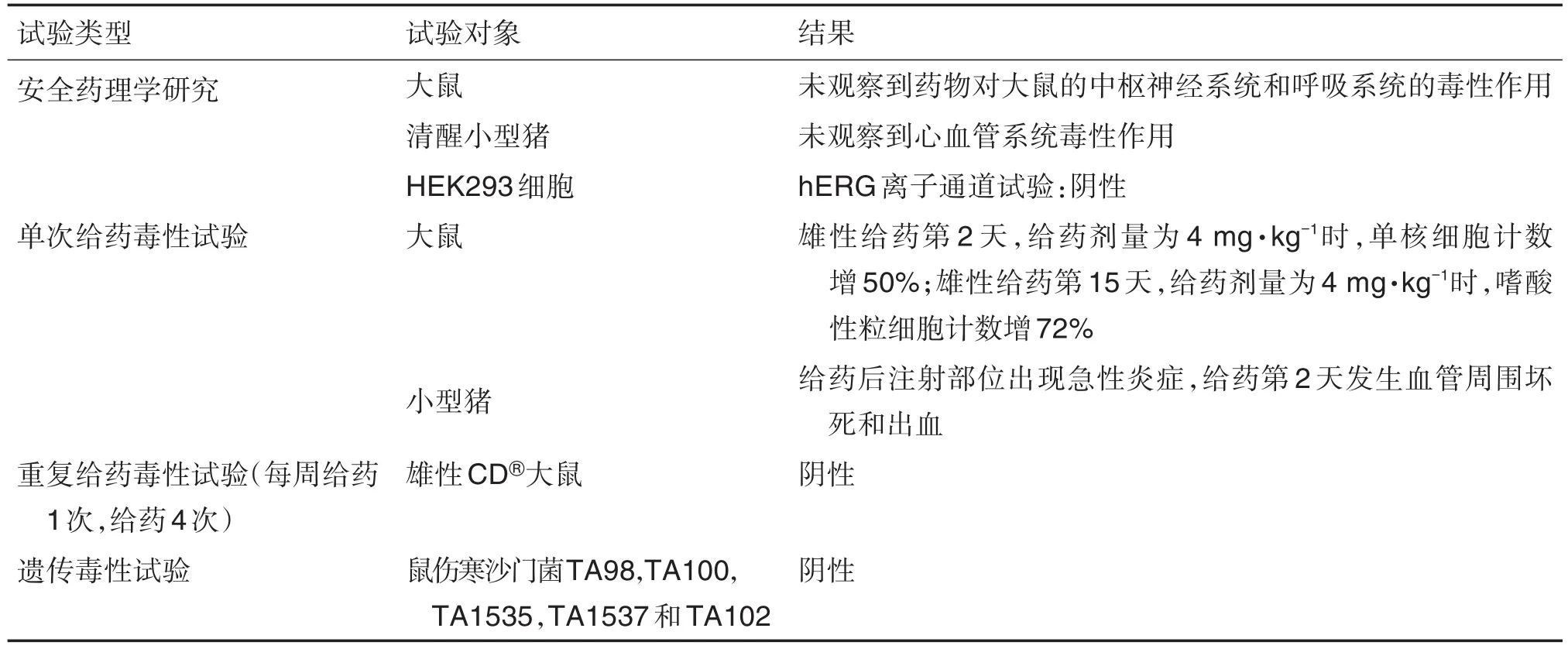
表4 Pluvicto非临床毒性试验及结果[42]
Pluvicto在非临床安全性评价中相继开展了不同动物种属安全药理试验、单次给药毒性试验、重复给药毒性试验和遗传毒性试验,目的在于发现Pluvicto潜在毒性。安全药理和单次给药毒性试验均采用不具有放射性的受试物PSMA-617和药物177Lu-PSMA-617进行。结果表明,多肽配体和放射性治疗药物整体对大鼠或小型猪的CNS、呼吸系统和心血管系统均未产生明显不良反应。单次给药毒性试验中体重、摄食量、血清生化、尿检、眼检、心电图、大体病理、组织病理学、器官重量等指标均未出现异常。仅对未标记的PSMA-617开展了重复给药毒性试验和遗传毒性试验,试验结果也均显示阴性,表明PMSA产生毒性的风险较低。根据美国FDA的《放射性肿瘤治疗药物:非临床研究和标签推荐工业指南》,人用药品177Lu-PSMA-617本身具有放射性,辐射会致癌、致畸、致突变,因此在非临床安全性评价中均未开展任何药物相关的其他遗传毒性、致癌性和生殖与发育毒性试验。
4 结语
自2018年以来,放射性治疗药物在肿瘤治疗领域取得了里程碑式突破,成为公认的最具潜力、效果最好的核素肿瘤治疗药物,广泛用于治疗癌症骨转移、前列腺癌、肝癌和甲状腺癌等,且治疗潜力巨大,其不断研发将满足未来集核素诊疗一体化的临床需求,推动放射性药物行业高质量发展。非临床安全性评价作为药物研发的重要一环,对预测放射性治疗药物的成药性及安全性具有重大价值,但目前国内尚无放射性治疗药物指导原则,非临床研究积累的经验不足,更多放射性治疗药物的研发也给安全性评价带来挑战。在制定放射性治疗药物的非临床安全性评价策略时,需遵循具体问题具体分析的原则,基于其特殊性进行有效性和安全性的综合分析,为其临床试验申请或上市申请提供非临床数据支持。
猜你喜欢
农村青少年科学探究(2022年4期)2022-07-29
当代水产(2021年6期)2021-08-13
现代临床医学(2021年3期)2021-07-16
工业催化(2020年9期)2020-11-13
无机化学学报(2020年7期)2020-07-20
小哥白尼(野生动物)(2019年5期)2019-08-27
中成药(2018年11期)2018-11-24
无机化学学报(2018年8期)2018-08-01
太空探索(2015年10期)2015-07-18
