
基于AhR信号通路的抗雌激素效应及其机制研究进展
2022-01-20 03:10谢昕岑于淼汝少国
生态毒理学报 2021年5期
谢昕岑,于淼,汝少国,*
1. 中国海洋大学海洋生命学院,青岛 266003 2. 廊坊师范学院,廊坊 065000
随着环境污染的日益加剧,外源化合物对生物体生殖健康的危害逐渐受到人们的关注。相较于具有雌激素活性的污染物,抗雌激素类物质的研究目前还处于起步阶段,且大多集中于雌激素(E2)结构类似物上。这些物质由于在化学结构上与E2具有一定的相似性,因此能够与E2竞争雌激素受体(ER)的配体结合域。但是,这类物质与ER结合后却无法发挥正常的雌激素效应,例如ICI 182780与ER结合后,会阻碍ER二聚体化,加速其降解;他莫昔芬(TAM)与ER结合后导致ER构象改变无法结合转录共激活因子,从而无法启动E2效应基因的转录。而对于那些化学结构与E2截然不同的具有抗雌激素活性的化合物,现有的报道多从下丘脑-垂体-性腺(HPG)轴的角度研究其对E2水平的影响,而缺乏对其触发分子起始事件的探究。由于生物体内的调控网络错综复杂,ER信号通路在体内并不是孤立存在的,许多调控因子或信号通路都可以与其产生交互作用,削弱或激活ER通路的作用效果。近期的研究发现,在外源化合物代谢过程中起着重要作用的芳香烃受体(AhR)途径若被激活,其将会通过多种途径干扰ER通路,表现出抗雌激素效应,这为我们研究外源污染物的抗雌激素效应机制提供了一个新的思路。本文通过查阅国内外相关研究文献,从AhR信号通路被外源配体激活后降低雌性个体E2水平及拮抗ER功能两方面系统论述AhR通路干扰ER通路的分子机制,以期为进行相关物质抗雌激素效应机制的研究提供借鉴与参考。
1 AhR-ER交互作用中相关的信号通路(Signaling pathways involved in AhR-ER crosstalk)
1.1 ER信号通路(ER signaling pathway)
ER属于类固醇/甲状腺激素核受体超家族,主要由3个功能性结构域组成(图1):NH2-末端结构域包含非配体依赖性的转录激活域(AF-1)[1-2],该结构域介导蛋白之间的相互作用[3-4]和目的基因转录激活;C结构域在受体二聚化及其与特异DNA序列结合过程中发挥着至关重要作用[5-6];D/E/F结构域则主要负责介导配体结合、受体二聚化、核转运以及靶基因的反式激活等分子事件[7-8],位于D/E/F结构域的配体结合域还包含着一个转录激活域(AF-2),与AF-1不同的是,AF-2结构和功能受到配体调控[9-11]。

图1 雌激素受体(ER)的二级结构(a)、ER的DNA结合域结合至靶基因启动子上雌激素受体响应元件(ERE)的 三维结构(b)和ER的配体结合域三维结构(c)Fig. 1 The secondary structure of estrogen receptor (ER) (a), three-dimensional structure of DNA binding domain of ER binding to estrogen receptor response element (ERE) on the target gene promoter (b) and three-dimensional structure of ligand binding domain of ER (c)
在没有配体的情况下,ER以非活化形式存在于细胞核中,此时“潜伏状态”的ER与共抑制因子热激蛋白90(HSP90)相互作用,ER转录活性被抑制[12]。在经典ER信号通路中ER由E2激活后,ER与抑制因子解离,并与p160类辅激活因子产生相互作用,目前已证明激活因子会通过组蛋白乙酰化作用增加染色质与ER及其他转录因子的作用,并增强受体上AF-1/AF-2的协同作用[13-14],随后形成的E2-ER复合体进入细胞核,并形成同源二聚体与靶基因DNA启动子区的雌激素受体响应元件(ERE)相结合,然后招募多种共调控因子和蛋白,启动或抑制下游基因的表达(图2A)[15-16]。除此之外,E2还可以通过非经典基因组模式(图2B)、膜受体介导的基因组模式(图2C)以及跨膜信号转导机制产生的快速基因组效应(图2D)发挥其生理功能[17-19]。
1.2 AhR信号通路(AhR signaling pathway)
AhR(图3)是一种配体依赖型转录因子,属于碱性螺旋-环-螺旋(bHLH)/Per-Arnt-Sim同源域(PAS)家族[20],在许多生物体内调节基因的表达。常见的AhR配体包括环境污染物如卤代芳烃及其类似物、生物体内的色氨酸及其代谢产物[21-24]。在未与配体结合的情况下,大部分AhR位于细胞质中,与HSP90二聚体及乙型肝炎病毒X蛋白结合蛋白2(XAP2)[25]结合,形成复合物。HSP90参与新合成AhR的折叠,能够稳定AhR的构象,使AhR维持在一种非活化的、可与配体结合的状态;XAP2一方面将AhR锚定在细胞骨架上,阻止AhR被入核蛋白importinβ识别,从而将其定位于细胞质中,另一方面这些分子伴侣能够抑制AhR的泛素化降解,从而使胞质中AhR的数量维持在一定水平[26]。当AhR配体进入细胞与AhR结合后,被激活的AhR会发生构象改变使其上的核定位序列暴露,随后AhR与上述分子伴侣解离并移位进入细胞核[27-28]。活化的配体-AhR将会在细胞核内与芳香烃受体核转运蛋白(ARNT)(图3)结合形成配体-AhR-ARNT复合体[29-30],并结合至特定的DNA识别位点cyp1a1及其他AhR效应基因上游启动子区的二噁英/外源物质反应元件(DREs/XREs)(核心五核苷酸序列:5’-GCGTG-3’),进而激活相关效应基因的转录(图4)[31-32]。
AhR在生物体内发挥着重要作用,其最为人们熟知的功能是AhR效应基因所编码的蛋白在外源物质(如2,3,7,8-四氯-二苯并-对-二噁英,简称TCDD)的体内代谢过程中发挥着至关重要的作用[33]。近期的研究表明,在无外源配体的情况下,AhR本身也参与调控生物体的诸多生理过程,如雌性个体的生殖。有证据表明,与正常小鼠相比,ahr缺陷型小鼠的繁殖效率显著降低,子代数量明显减少[34]。而当AhR通路被外源配体激活后,也将通过多种途径干扰ER通路,发挥抗雌激素效应。
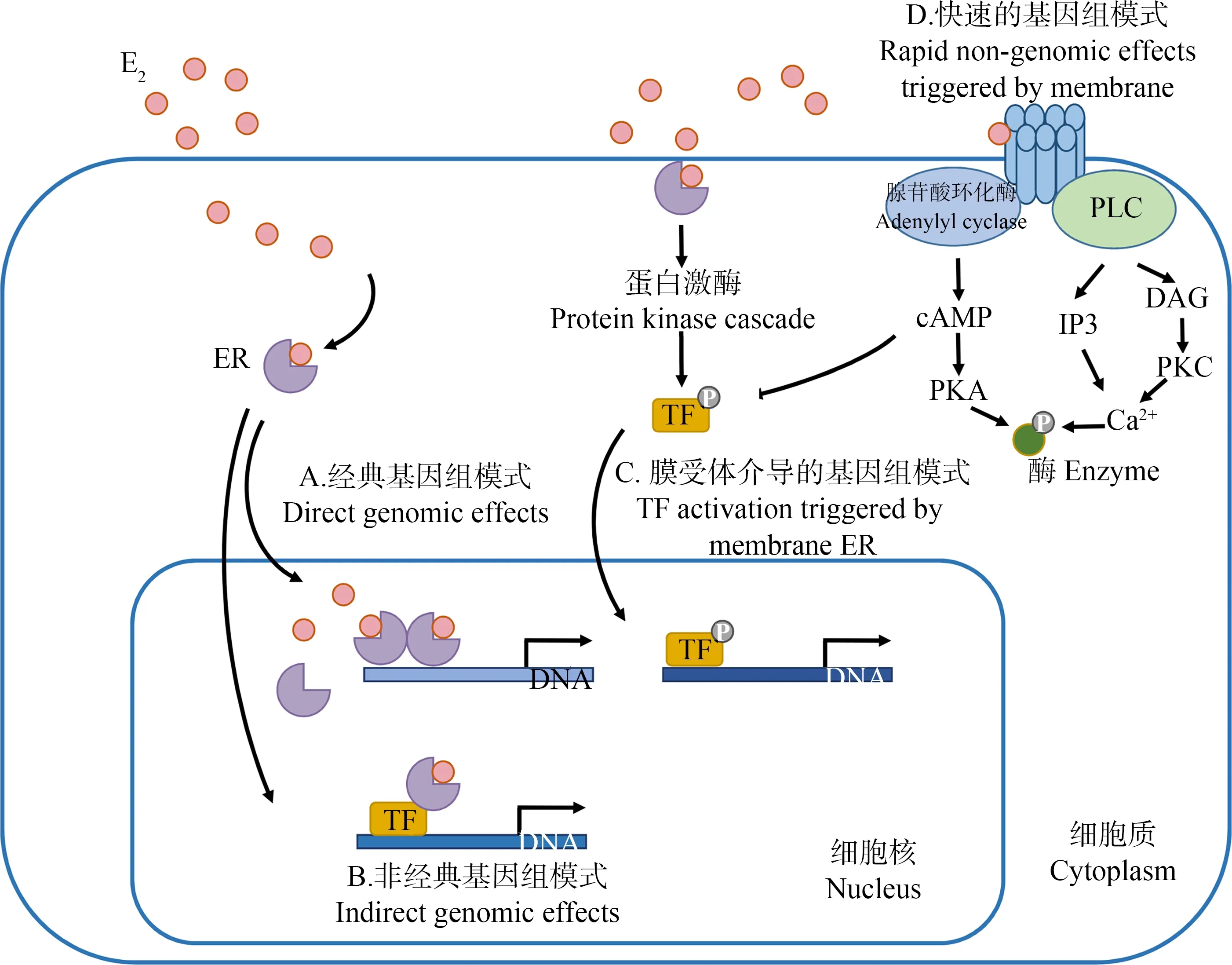
图2 雌激素(E2)在细胞内的作用模式注:TF为转录因子;cAMP为环磷酸腺苷;PKA为蛋白激酶A;PLC为磷脂酶C;IP3为1,4,5-三磷酸肌醇;DAG为二酰甘油;PKC为蛋白激酶C。Fig. 2 Mechanisms involved in the cellular actions of estrogen (E2)Note: TF is transcription factor; cAMP is cyclic adenosine monophosphate; PKA is protein kinase A; PLC is phospholipase C; IP3 is inositol 1,4,5-triphosphate; DAG is diacylglycerol; PKC is protein kinase C.

图3 芳香烃受体(AhR)和芳香烃受体核转运蛋白(ARNT)的二级结构注:bHLH为碱性螺旋-环-螺旋结构,PAS A和PAS B为Per-Arnt-Sim同源域。Fig. 3 The secondary structure of aryl hydrocarbon receptor (AhR) and aryl hydrocarbon receptor nuclear translocator (ARNT)Note: bHLH is basic helix-loop-helix domain, PAS A and PAS B are Per-Arnt-Sim domains.
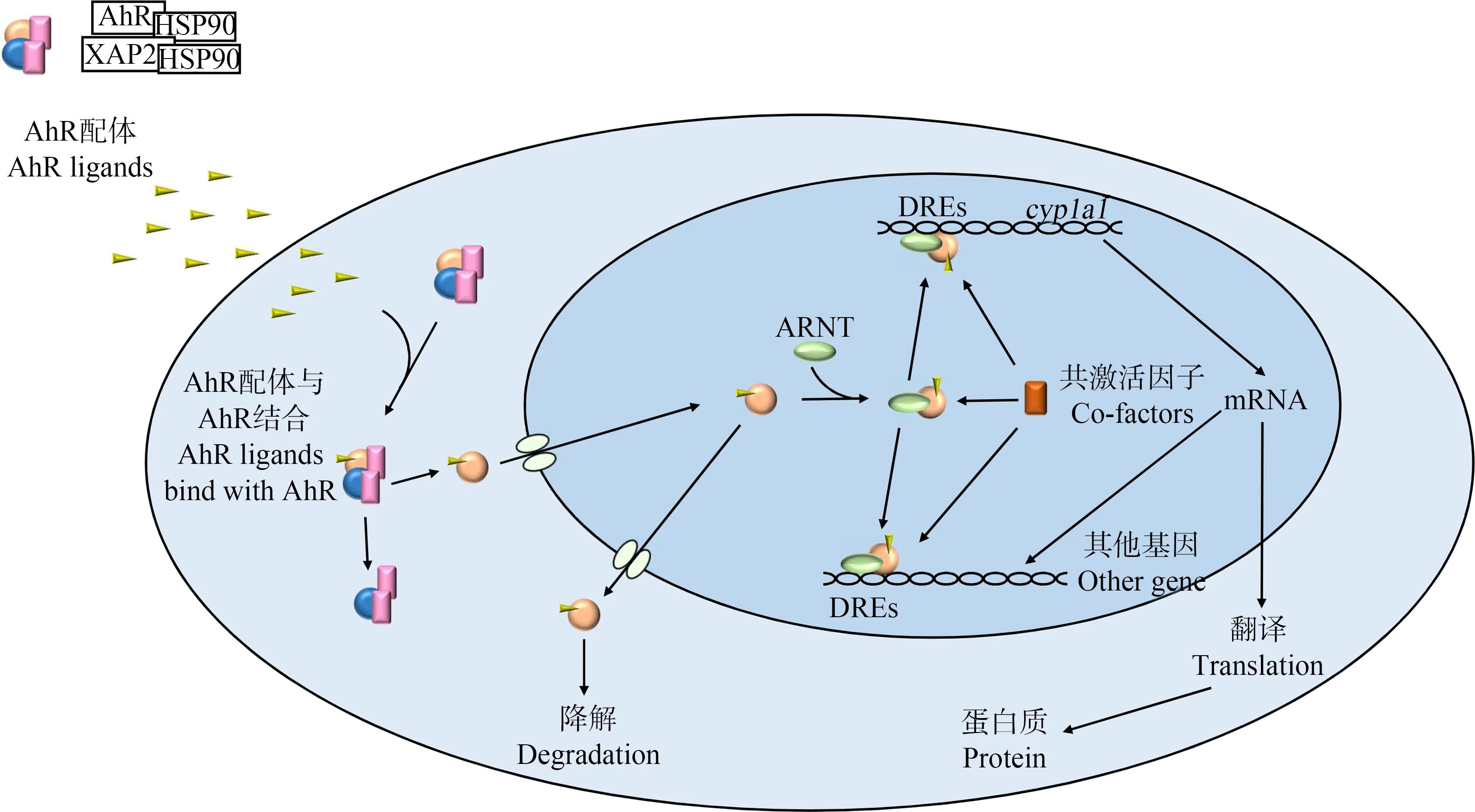
图4 AhR信号通路注:DREs为二噁英/外源物质反应元件,HSP90为热激蛋白90,XAP2为乙型肝炎病毒X蛋白结合蛋白2。Fig. 4 AhR signaling pathwayNote: DREs are dioxin response elements; HSP90 is heat shock protein 90; XAP2 is hepatitis B virus X protein-associated protein 2.
2 基于AhR信号通路的抗雌激素效应分子机制(Molecular mechanisms of anti-estrogenic effects based on AhR signaling pathway)
目前已有多项研究证实AhR激动剂在不同生物学层次上均发挥着抗雌激素效应。首先E2效应基因转录水平的变化是检测外源污染物抗雌激素效应最为快速且综合的指标,如Bemanian等[35]发现TCDD能够抑制E2对大西洋鲑(Salmosalar)esr1和vtg转录的激活效应;AhR的另一激动剂2-苯基色原酮也能够显著降低MCF-7细胞中E2效应基因pr和pS2的转录水平[36];Wang等[37]利用报告基因体外转染MCF-7细胞,发现E2能显著上调含有vtgA2、pS2和组织蛋白酶D(cathepsin D)启动子区域的报告基因的mRNA表达水平,而TCDD则能够使E2的对上述基因的转录激活作用消失,类似结果也出在T47D细胞系中。除E2效应基因的转录水平受到影响外,其蛋白水平也可被AhR激动剂改变。如TCDD、多氯联苯126、β-萘黄酮、苯并芘和二吲哚甲烷均能够显著降低鲤鱼(Cyprinuscarpio)肝脏细胞中卵黄原蛋白(VTG)的表达水平[38],VTG是卵细胞成熟的关键物质,其蛋白水平降低将影响雌鱼生殖。Oenga等[39]的研究还证实TCDD和多氯联苯(PCBs)能够削弱E2对MCF-7细胞体外增殖的诱导作用,TCDD和E2联合染毒切除卵巢的小鼠,发现TCDD能够抑制外源性E2诱导的子宫湿质量增加[40-41];有机氯杀虫剂甲氧氯可通过激活AhR通路抑制体外小鼠卵巢窦卵泡的生长并诱导闭锁卵泡生成,当敲除ahr后该抑制作用则不复存在[42]。上述众多研究从基因转录水平、蛋白水平、细胞水平以及组织水平证实了AhR激动剂的抗雌激素效应,而这种效应的产生源于被激活的AhR通路与ER通路之间的种种交互作用。尽管二者之间详细的分子机制还有待进一步研究,从目前的研究结果来看,可将其归纳为以下5个方面(图5)。
2.1 降低E2水平(Reducing the level of E2)
2.1.1 抑制E2合成(Inhibition of E2synthesis)
在性成熟的雌性个体中,胆固醇在一系列酶的作用下合成E2,这些酶包括胆固醇侧链裂解酶(CYP11A)、3β-羟类固醇脱氢酶(3β-HSD)、17α羟化酶/C17,20-裂解酶(CYP17)、17β-羟类固醇脱氢酶(17β-HSD)和芳香化酶(CYP19)等(图6),其表达水平或活性的变化会直接影响雌性生物体内的E2水平。

图5 AhR信号通路干扰ER信号通路的机制注:iDRE为二噁英/外源物质反应元件类似序列,p300、HSP90和RIP140为转录因子。Fig. 5 Mechanisms of AhR signaling pathway interfering with ER signaling pathwayNote: iDRE is similar sequence of dioxin response element; p300, HSP90, RIP140 are transcription factors.
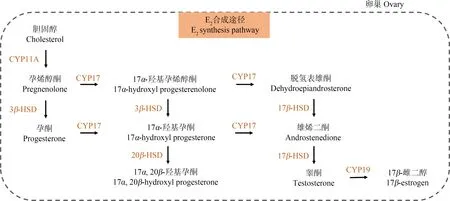
图6 E2合成途径Fig. 6 E2 synthesis pathway
在E2生物合成的最后一步,cyp19编码的芳香化酶将睾酮(T)转化成E2,被认为是该生物学过程的限速步骤,芳香化酶CYP19表达量和活性的变化可显著改变E2的合成量。在硬骨鱼大脑中,cyp19b是编码芳香化酶的主要基因[43-45],同时其本身还是经典的E2效应基因[46],Cheshenko等[47]研究发现TCDD暴露会降低斑马鱼体内cyp19b的表达水平。体外实验结果显示TCDD还会抑制zfcyp19b荧光素酶报告基因对E2响应的灵敏度,而加入AhR拮抗剂可以部分(体内)或完全(体外)缓解这种效应,证实了TCDD对cyp19b表达的抑制依赖于AhR。
2.1.2 促进E2代谢(Enhanced metabolism of E2)
AhR激动剂还可通过上调E2代谢相关酶的活性,促进E2代谢,进而降低内源E2水平。E2的代谢主要发生在肝脏中,第一步是肝脏细胞色素(CYP)P450催化,NADPH参与的氧化代谢[48]。由AhR通路的效应基因所编码的CYP1家族在哺乳动物内源E2的代谢过程中发挥着至关重要的作用,CYP1和CYP3A催化E2产生2-OHE2、少量的4-OHE2及其他羟基代E2[49],2-OHE2和4-OHE2由儿茶酚-O-甲基转移酶(COMT)进一步代谢成2-MeOE2和4-MeOE2[50]。TCDD等一些AhR激动剂能够诱导乳腺癌细胞MCF-7、T47D、MDA-MB-231中cyp1a1和cyp1b1的表达,增强E2代谢,进而降低E2在细胞内的水平并抑制细胞增殖[51-54];Lu等[55]的研究采用高效液相色谱(HPLC)检测了TCDD对MCF-10F细胞中E2及其代谢物产物水平的影响,结果显示E2水平降低,4-OHE2含量增多,表明TCDD促进了细胞中E2的代谢。
2.2 拮抗ER功能(Antagonizing the functions of ER)
E2水平降低势必会表现出抗雌激素效应,然而有些AhR激动剂并不能影响生物体或细胞内E2合成或代谢途径,却仍削弱E2的生理功能。例如Helle等[56]研究发现3-甲基胆蒽(3-MC)能够抑制雌性小鼠子宫内E2效应基因的转录水平,然而作者利用3-MC体外暴露雌性大鼠肝脏微粒体,发现微粒体E2水平与对照组无明显差异,E2代谢物水平也没有发生变化。可见3-MC并非通过降低E2水平发挥其抗雌激素效应,拮抗ER功能可能是其产生抗雌激素效应的重要途径。
2.2.1 直接抑制雌激素效应基因的转录(Direct inhibition of estrogen-regulated gene transcription)
受体被激活后往往通过与靶基因启动子区特定的序列结合,激活基因表达。同属核受体转录因子家族的AhR和ER有各自特定的识别序列——DRE和ERE。然而研究发现,在一些E2效应基因启动子区ERE附近存在着DRE或DRE类似序列(iDRE),当AhR受到配体激活后,活化的AhR复合体可能会与此处的DRE/iDRE结合,这将会占据ER复合体的结合空间,阻碍ER复合体与ERE结合,从而抑制雌激素效应基因的转录。目前已在cathepsinD[57-59]、ps2[60]、hsp27[61]和c-fos[61]等雌激素效应基因的启动子区发现了功能性iDRE(图7),但其作用机制存在显著差异。例如cathepsinD启动子的近端调控区(转录起始位点前250 bp附近)含有多个ERE,包括可与ERα/Sp1结合的GC(N)19ERE(ERE1/2)功能域,可与ERα/Sp1结合的高GC功能域以及可与USF1/2-ERα复合物结合的E-box功能域,在体外构建GC(N)19ERE1/2介导的报告基因体系,发现E2能够诱导该报告基因的表达,而TCDD能够抵消这种诱导作用[57]。对GC(N)19ERE1/2功能域附近(-195至-165)进行序列分析可知,cathepsinD启动子区的-177到-181为GCGTG,是AhR复合体识别DRE的核心五核苷酸,将该处iDRE突变后,TCDD对E2的拮抗效应随之消失,后续的凝胶阻滞实验表明AhR复合物能够与该iDRE相结合,继而阻止ERα/Sp1与GC(N)19ERE1/2的正常结合,抑制了E2介导的反式激活[57-59]。

图7 cathepsin D、c-fos、hsp27和ps2基因启动子区功能性外源物质反应元件类似序列(iDRE)的示意图[89]Fig. 7 Functional xenobiotic response elements (iDRE) in promoters of the cathepsin D, c-fos, hsp27, and ps2 genes [89]
2.2.2 与ER信号通路竞争共调控因子(Competition for cofactors)
受体调控靶基因表达除了需要配体外,还需要多种共调控因子(cofactors)的共同参与。ER上AF-1和AF-2的转录活性依赖于共调控因子的相互作用,这些共调控因子分为共激活因子(coactivators)和共抑制因子(corepressors)[62]。
AhR信号通路与ER信号通路同为核受体通路,被激活时将会竞争共用的转录因子,互相产生干扰。如Ricci等[63]将包含人类cyp1a1基因启动子序列(-1612/+292,含至少3个功能性DRE)的报告基因导入ECC-1细胞中,并用E2和TCDD联合暴露该转染细胞,结果显示联合暴露组中的荧光素酶活性较TCDD单独暴露组降低了74%,表明E2抑制了TCDD对cyp1a1的转录激活作用,但E2对TCDD诱导的cyp1b1的转录水平无显著影响。作者进一步研究发现,cyp1a1的转录受转录因子NF-1的调控,而NF-1不调控cyp1b1的表达[63];同时NF-1是E2效应基因转录中必不可少的转录因子,因此推测2条通路间的拮抗关系可能源于它们竞争细胞内有限数量的NF-1。作者进一步向该转染细胞中导入过量NF-1表达质粒后,缓解了E2对cyp1a1的转录抑制作用,证实了上述推论。此外,AhR信号通路中关键因子ARNT也在ERα和ERβ诱导的转录激活中发挥着激活因子的作用[64],当AhR通路被激活后大量的ARNT将被招募至AhR通路中,从而减少ER通路中可利用的ARNT,产生抗雌激素效应。例如Rüegg等[65]的研究显示含3xERE荧光素酶报告基因的HC11细胞(HC11-ERE)暴露于TCDD,形成大量AhR/ARNT复合物,导致荧光强度减弱。
2.2.3 促进ER降解(Enhanced metabolism of ER)
在细胞中,蛋白质降解主要有2种途径:溶酶体途径和泛素-蛋白酶体途径。其中泛素化在蛋白质特异性降解过程中发挥至关重要的作用[66]。在泛素-蛋白酶体途径调控的蛋白质降解中,E3泛素连接酶决定了降解目标的特异性[67-69]。有研究表明,AhR除了具有转录因子活性外,其被相应的配体激活后还是一种E3泛素连接酶,能够对ERα进行泛素化修饰,从而促进ERα的降解。(1)在体外实验中,配体-AhR复合体以一种依赖于泛素激活酶E1/泛素结合酶E2的方式表现出了自身泛素化的活性。(2)被3-MC活化的AhR能够识别ER,然后诱导ER的泛素化[70]。(3)ER的降解会加速AhR自身的降解,这是E3泛素连接酶自身泛素化的一个典型特征[71]。上述结果均表明,AhR在E3泛素连接酶复合体中可能作为特异性识别底物的接头蛋白,其功能与SCF复合体中F-box蛋白的功能相似[68, 71]。Yanagisawa等[72]已在Hela细胞中纯化出了含有AhR的泛素连接酶复合体,该复合物由受损DNA结合蛋白1(DDB1)、AhR、ARNT、转导素-β样蛋白3(TBL3)、滞蛋白(CUL4B,泛素连接酶的核心成分)和环状结构蛋白1(Rbx1)组成。另一项研究表明,AhR配体能够诱导小鼠子宫中ERα蛋白的降解,然而在ahr缺陷型小鼠中,用AhR配体处理则不会改变ERα的蛋白表达水平[73-74];在ERα阳性乳腺癌细胞MCF-7、T47D和ZR75-1中,AhR在其配体TCDD的参与下与ERα结合,并以不依赖激素的方式促进ERα通过泛素-蛋白酶体途径降解[75]。
3 展望(Expectation)
AhR作为一种配体激活的核转录因子,在人体中发挥着重要作用,它既能够与有毒物质结合参与生物体解毒过程,也能够调节雌性生物体的生殖[76]。
AhR激动剂广泛存在于环境中,例如近年来针对我国三峡水库的化学分析研究显示,水库沉积物被持久性有机污染物(POPs)所污染,包括PCBs、有机氯杀虫剂(OCPs)以及多环芳烃(PAHs)[77-79],其中许多化合物都能激活AhR,将对水生生物乃至人类造成严重危害;韩国始华湖[80]及蔚山湾[81]沉积物中也有各类AhR激动剂检出。各类化合物的联合作用诱导AhR的效力极大增加,因此鉴定并识别出混合物中AhR激动剂的不同组分及含量,能够更好地为环境污染物优先级的评定及管制办法的制定提供参考。目前已开发出许多离体方法,如信号通路分析、报告基因等用于分析外源化合物的潜在影响,但在处理数以万计化合物时成本高昂且耗时费力[82]。出于上述原因,计算机技术正在迅速发展。基于定量构效关系(QSAR)建模预测化合物的毒理学效应,具有耗时少、成本低的优势[83],因此可以作为监测未来环境中AhR激动剂的重要手段。如Xiao等[84]就首先进行毒理学实验鉴定出三峡水库沉积物中潜在的AhR激动剂,接着在此基础上使用QSAR建立数学模型,根据化合物分子结构性质数据模拟以预测筛选出的AhR激动剂的生物活性,确定了沉积物中新的潜在AhR激动剂——苯并噻唑和2-巯基苯并噻唑。
此外,AhR通路作为当前抗雌激素效应研究热点,探明其发挥抗雌激素效应的机制不仅有助于AhR激动剂的毒性效应预测,更能够以此作为激素依赖性肿瘤的治疗靶点。流行病学研究发现意外暴露于TCDD的妇女,其乳腺癌和子宫内膜癌的发病率较低[85];AhR的配体二吲哚甲烷(DIM)类似物[86]是吲哚-3-甲醇(I3C)在酸催化下形成的二聚产物,相对于TCDD等化合物毒性较低,DIM(1 mg kg-1)暴露24 h对雌性大鼠肿瘤生长有显著抑制作用[87];AhR内源性配体2-(1’-吲哚-3’-羰基)-噻唑-4-羧酸甲酯(ITE)能抑制离体子宫内膜癌(EC)细胞的增殖和迁移,在小鼠体内也能抑制EC细胞的异种移植生长[88]。上述结果均提示AhR通路可能是开发治疗乳腺癌和子宫内膜癌药物的重要靶点,因此开发或合成具有较低毒性、较高抗雌激素和抗肿瘤活性的AhR外源性配体,还可将AhR作为激素依赖性肿瘤治疗的关键中间物质,应用于乳腺癌的预防和治疗。
猜你喜欢
生物化学与生物物理进展(2022年8期)2022-08-20
食品安全导刊(2021年20期)2021-08-30
中华养生保健(2020年9期)2021-01-18
无机化学学报(2020年7期)2020-07-20
渤海大学学报(自然科学版)(2020年3期)2020-02-02
中国脑血管病杂志(2019年8期)2019-03-13
天然产物研究与开发(2018年6期)2018-07-09
科技创新导报(2016年30期)2017-03-15
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
