
·Cl引发萘的大气氧化机制及动力学
2022-01-20 03:09陈杰马芳芳郭熙瑞谢宏彬
生态毒理学报 2021年5期
陈杰,马芳芳,郭熙瑞,谢宏彬
大连理工大学环境学院,工业生态与环境工程教育部重点实验室,大连 116024
氯自由基(·Cl)具有强的化学反应活性,与大气中大多数污染物的反应速率常数(kCl)比羟基自由基(·OH)对应的反应速率常数(kOH)高10倍~100倍[1-4]。过去·Cl被认为主要分布于沿海地区,由海盐经多相化学反应生成,浓度可达·OH的1%~10%[1,5-7]。因此,一直认为·Cl对沿海地区污染物的转化起着重要的作用[8-9]。然而,最近几年在中国、美国和加拿大等国的内陆城郊区域发现较高浓度的·Cl前体物质ClNO2[10-13]。这表明·Cl对有机污染物的转化可能起着比以前更为重要的作用。近期关于北京地区大气污染的一项研究指出,·Cl对烷烃类污染物的转化贡献率高于·OH[14]。此外,值得指出的是,·Cl引发有机污染物的转化可能导致与·OH引发反应不同的大气转化机制,进而可能会导致不同的大气归趋[3,15-17]。因此,·Cl引发城市大气有机污染物的转化亟待研究。
多环芳烃(polycyclic aromatic hydrocarbons, PAHs)是影响城市大气环境质量的重要污染物,主要由化石燃料和生物质燃烧排放,组成成分复杂[18-20]。萘(naphthalene, Nap)是分子量最小、挥发性最强的PAHs。此外,Nap还是重要的化学品,被用作化工原料[21]。较高的挥发性使Nap能在生产、运输和使用过程中进入大气环境。全球尺度的大气模型模拟结果指出,Nap的含量超过PAHs总量的2/5[18]。特别是在洛杉矶,Reisen和Arey[19]检测到Nap的大气浓度高达1 600 ng·m-3。重要的是,前人研究发现,Nap还是二次有机气溶胶(secondary organic aerosol, SOA)的重要前体物[22-24]。如Huang等[23]研究发现,在北京地区的雾霾事件中,Nap及Nap的衍生物转化生成的SOA最高浓度可达到4.6~6.8 μg·m-3,约贡献了SOA总量的14.9%。考虑到Nap是一种重要化学品及其大气化学的重要性,有必要研究Nap的大气转化。
日间被大气中的·OH和·Cl氧化是Nap的重要大气归趋[22,24-27]。前人采用实验和理论计算的方法研究了·OH引发Nap的大气氧化机制与动力学[26,28-29]。研究指出,·OH主要加成到Nap分子上形成加成中间体Nap-OH,随后与O2反应生成萘酚和过氧自由基(RO2·)。RO2·既可以经单分子异构化生成肉桂醛,也可与NO反应生成烷氧自由基(RO·)和有机硝酸酯,RO·经环化后进入下一个O2/NO反应,最终生成含羰基的双环中间体[29]。然而,针对·Cl引发Nap的大气氧化,仅有一些实验测量了两者的反应速率常数并鉴别了部分氧化产物的碎片信息[25,30]。而对·Cl引发Nap的具体大气转化机制还不清楚。
近年来,随着量子化学计算方法计算精度的提高和计算机计算能力的大幅提升,量子化学方法已经成为研究化学品及大气污染物转化机制和动力学的重要工具,在大气化学及化学品风险评价中发挥着重要作用[31-32]。如大连理工大学陈景文团队近年来利用量子化学方法成功揭示了多种化学品及大气污染物的转化机制和动力学[32-35]。
本研究使用量子化学计算结合动力学模拟的方法研究了·Cl引发Nap的大气氧化机制与动力学,包括·Cl与Nap的引发反应及引发反应生成的重要中间体的后续反应(包括加成中间体与O2/NO的双分子及RO2·/RO·的解离和异构化)的转化机制与动力学。此外,使用化学品水生毒性评估软件ECOSAR评估了部分闭壳层产物的水生毒性[36]。
1 计算方法(Computational methods)
使用Gaussian 09软件包进行电子结构优化及能量计算[37]。在ωB97XD/6-31+G(d,p)计算水平下,对所有反应渠道涉及的反应物(R)、反应前络合物(RC)、过渡态(TS)、反应后络合物(PC)和产物(P)的结构进行优化[38]。在相同计算水平下,通过内禀反应坐标计算确定TS连接的R和P。使用ωB97XD/6-311++G(3df,2pd)方法计算单点能量。通过公式(1)计算每个物种的能量。
E=ESP+Ecorr
(1)
式中:ESP为单点能;Ecorr为ωB97XD/6-31+G(d,p)计算水平获得的零点矫正能。
本论文所有单分子反应和双分子反应速率常数均采用传统过渡态理论(canonical transition state theory, TST)进行计算,TST计算在MULTIWELL-2014.1软件包中的Thermo模块进行[39-42],使用Eckart约近考虑单双分子反应中H迁移和H夺取反应的隧道效应[41]。
2 结果与讨论(Results and discussion)
2.1 ·Cl引发反应
原则上,·Cl可以夺取Nap的H原子或·Cl加成到不饱和键上。Nap分子存在8个H原子和10个不饱和C原子(图1),因此,·Cl和Nap反应有H夺取和Cl加成2类,共18条可能的反应渠道。由于Nap具有D2h对称性,·Cl与Nap反应需要考虑的可能渠道可以降低到5条,即:·Cl夺取Nap的H12和H13;·Cl加成到Nap的C4、C5和C6位置(图1)。基于计算的热力学数据(表1)可知,·Cl加成到C5和C6位置放热显著高于H夺取反应。因此,从热力学上判断,加成反应更容易发生。此外,值得指出的是,前人研究也发现·Cl夺取sp2杂化的C上的H很难发生[43]。因此,本研究仅考虑·Cl和Nap的加成反应。
为确定·Cl和Nap反应的加成位点,首先,我们进行了二维势能面扫描[44]。以Nap分子C骨架平行且距离为0.25 nm的平面为扫描平面,以C3-C1和C3-C4方向分别为X轴和Y轴。·Cl的移动范围为X=0~0.26 nm和Y=0.07~0.24 nm所包含的矩形,该矩形能覆盖Nap的C4、C5和C6上方区域。·Cl在X和Y轴上的扫描步长均为0.01 nm,共得到约500个扫面点的能量数据,根据此能量数据绘制的二维扫描势能面如图2所示。由图2可知,X=0.1~0.26 nm和Y=0.16~0.24 nm构成的区域(C5、C6附近)的能量显著低于X=0~0.15 nm和Y=0.07~0.10 nm构成的区域(C4附近),即·Cl在Nap的C5和C6上部区域移动时,体系能量更低。结合前文未能优化到C4加成产物的稳定构型,说明C4不可能是主要的加成位点,加成反应可能的主要加成位点为C5和C6。
为了进一步确定加成到C5和C6哪个位置更可行,对·Cl加成到C5和C6位置进行一维势能面扫描。C-Cl垂直距离(rC-Cl)为0.19~0.28 nm,每隔0.005 nm进行扫描。·Cl加成到C5位置的扫描曲线如图3(a)所示,由图可知,当rC5-Cl在0.19~0.28 nm范围变化时,随着C5-Cl距离的逐渐增加,能量逐渐升高,说明·Cl加成到C5位置是无能垒的过程。·Cl
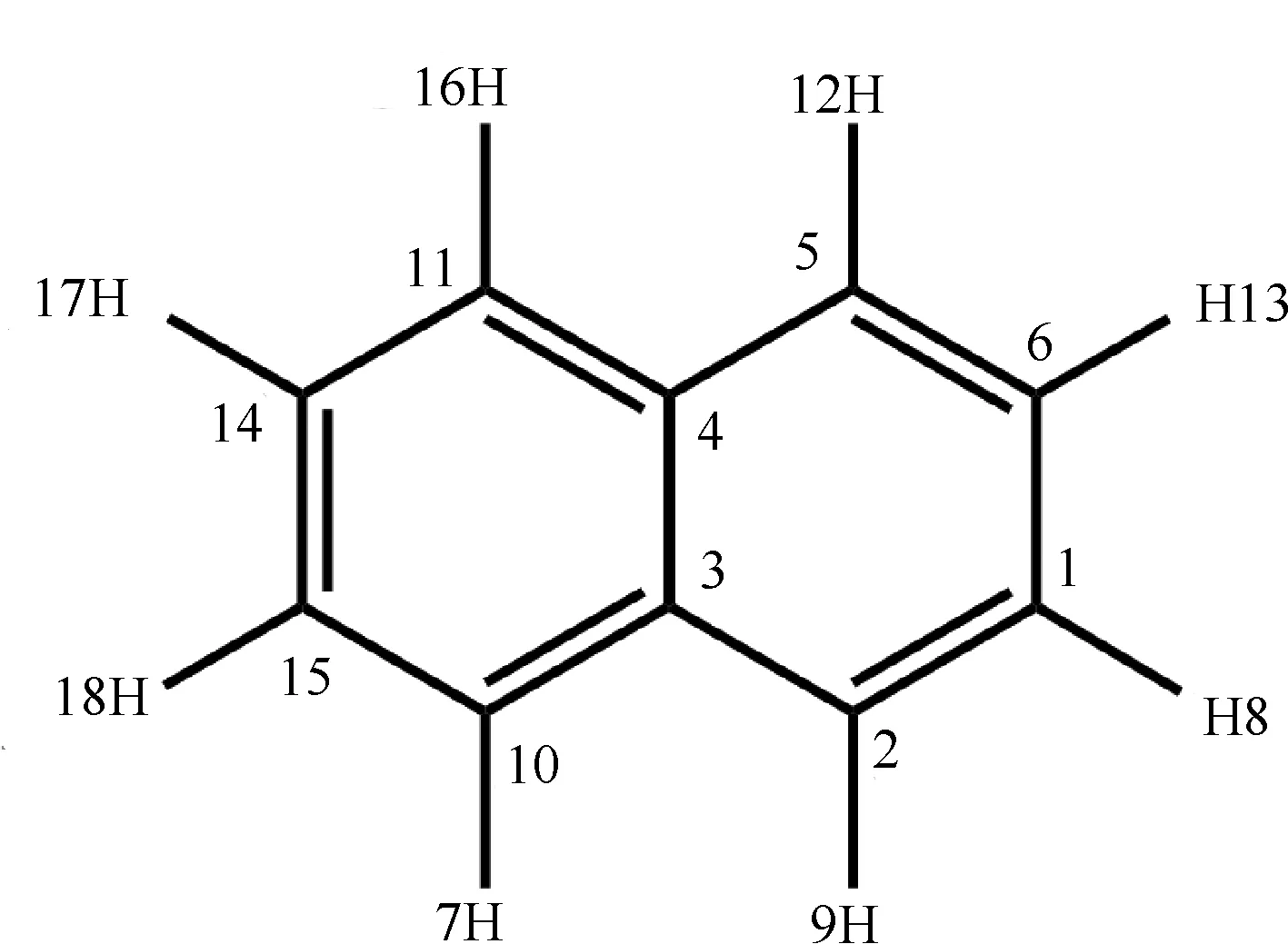
图1 萘(Nap)的分子结构及原子序号Fig. 1 Structure and atomic labeling of naphthalene (Nap)
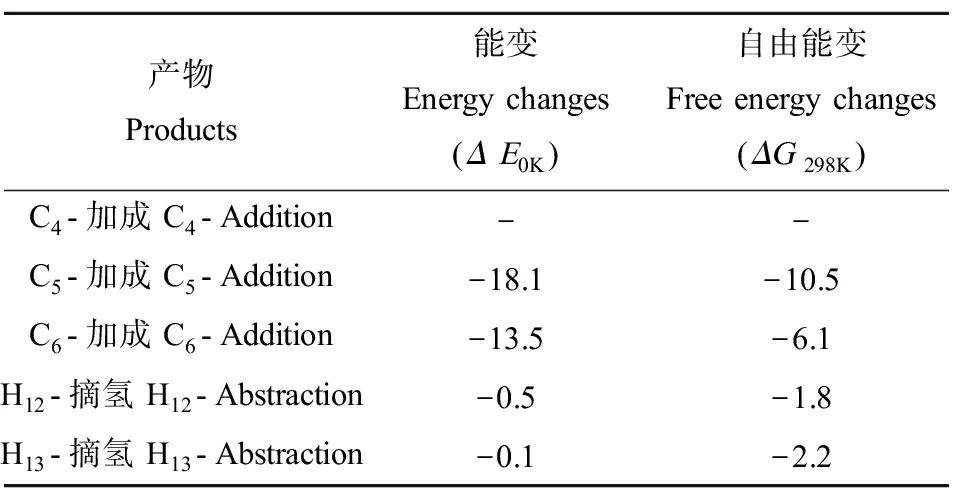
表1 在ωB97XD/6-311++G(3df,2pd)//ωB97XD/6-31+ G(d,p)计算水平下,·Cl和Nap反应的反应能Table 1 Calculated energy changes of ·Cl + Nap reaction at the B97XD/6-311++G(3df,2pd)//ω B97XD/6-31+G(d,p) level (kcal·mol-1)
加成到C6位置的扫描曲线如图3(b)所示,由图可知,在rC6-Cl=0.275 nm和rC6-Cl=0.19 nm时,·Cl和Nap已经形成2种不同的局域能量极小点,分别对应可能的C5和C6加成。因此,即使对·Cl加成到C6位置进行扫描,也会首先形成加成到C5位置的中间体,表明C5位置是最可行的加成位点,与热力学结果一致。值得指出的是,在·Cl与Nap的氧化实验中,·Cl加成到C5的加成中间体也被推测为主要加成产物,支持计算结果的可靠性[25]。接下来仅考虑C5位置加成中间体·C10H8Cl(R1)的后续反应。
2.2 R1与O2反应
在对流层大气环境中,和其他C中心自由基反应类似,加成中间体R1可能进一步与O2发生双分子反应[29,45]。R1与O2可以发生直接H夺取和O2加成2类反应:(1) O2直接夺取R1中—CClH—基团上的H生成C10H7Cl和HO2·;(2) O2通过顺式(syns)或反式(anti)2个方向加成到自旋密度较高的C6(0.59)、C2(0.45)、C10(0.14)、C14(0.16)和C4(0.13)位置形成过氧自由基中间体R1-iOO-s/a(i=2,4,6,10,14,s/a=syns/anti,O2与·Cl方向相同为syns加成,相反为anti加成)。表2中列出了R1与O2反应的能量信息。基于计算的反应能垒(ΔE0K≠)可知,相比于其他反应途径(直接H夺取和加成到C10、C14和C4位点),O2通过syns和anti加成到C2(ΔE0K≠=6.7 kcal·mol-1和5.4 kcal·mol-1)和C6(ΔE0K≠=8.6 kcal·mol-1和5.2 kcal·mol-1)位点形成R1-2OO-s/a和R1-6OO-s/a是最可行的。此外,通过对比加成渠道形成的加成中间体的稳定性(基于ΔE0K和ΔG298K判断)发现,相比于其他加成位点的加成产物,C2和C6加成产物更稳定,这主要是因为这些加成产物中保留着完整的苯环。O2与OH-Nap的反应中也发现了类似的现象[22]。因此,在下文中我们仅考虑O2加成到C2和C6位置形成的加成中间体R1-2OO-s/a和R1-6OO-s/a的反应。
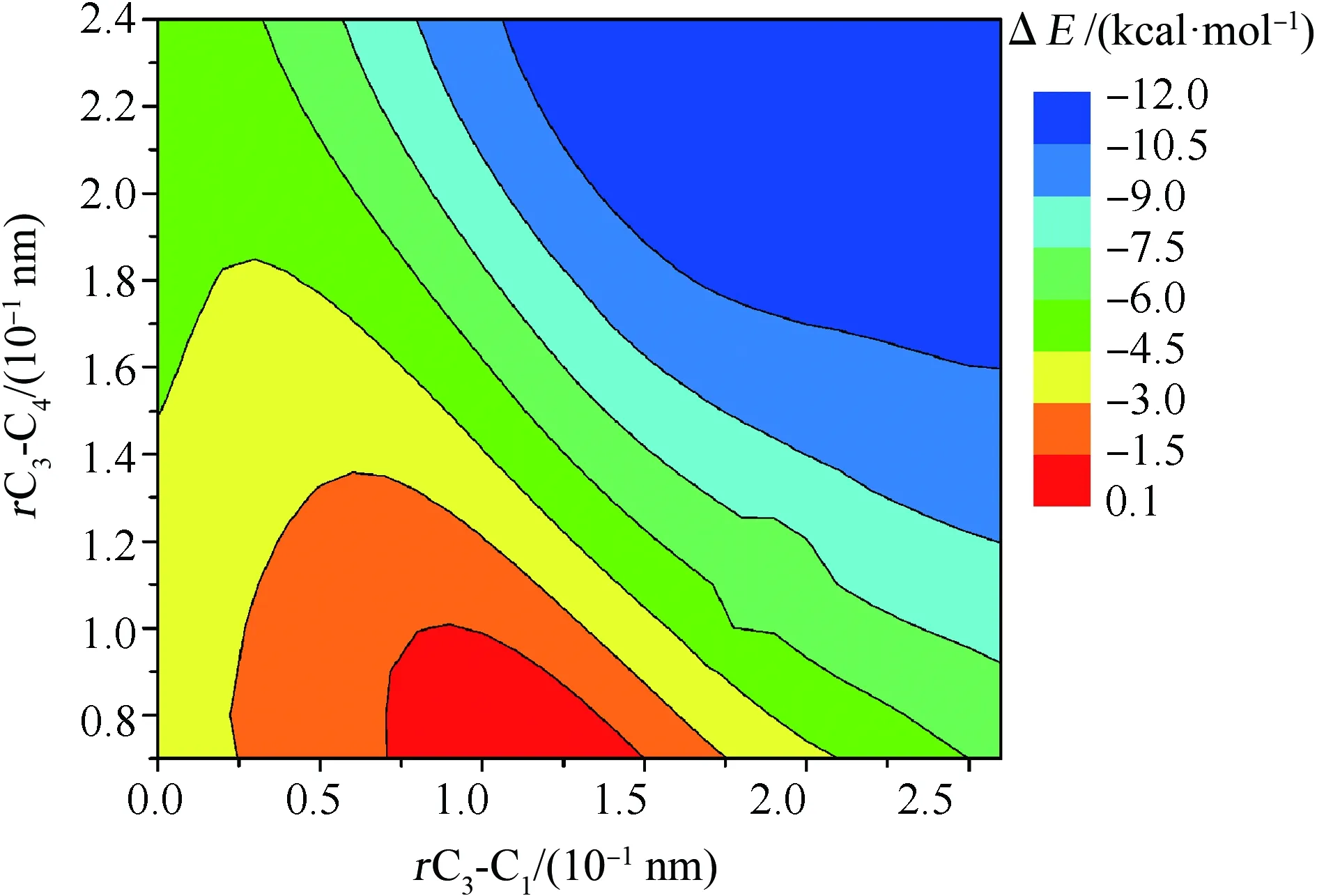
图2 在ωB97XD/6-31+G(d,p)计算水平下, ·Cl加成到Nap的二维扫描势能面(PES)注:ΔE为扫描点相对于反应物·Cl+Nap的能量,单位为kcal·mol-1。Fig. 2 Contour plots of the two-dimensional scanned potential energy surface (PES) for ·Cl addition to the Nap at the ωB97XD/6-31+G(d,p) levelNote: ΔE is the relative energy (kcal·mol-1) with reference to the reactants ·Cl+Nap.
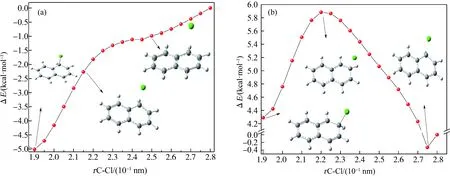
图3 在ωB97XD/6-31+G(d,p)计算水平下,·Cl加成到Nap的C5(a)和C6(b)位点的扫描势能曲线注:rC-Cl指C-Cl间距离,单位为10-1 nm;rC-Cl=0.28 nm构型的能量作为ΔE计算的参考点,单位为kcal·mol-1。Fig. 3 Scanned potential energy curves for ·Cl addition to C5(a) and C6(b) sites of Nap at the ωB97XD/6-31+G(d,p) levelNote: rC-Cl is the value of Cl-C distance ΔE is the relative energy (kcal·mol-1) with reference to the configuration (rC-Cl=0.28 nm).
2.3 R1-2OO-s/a和R1-6OO-s/a的反应
氧化加成中间体R1-2OO-s/a和R1-6OO-s/a能发生自身异构化反应或与大气中的NO/HO2·发生双分子反应[45-46]。R1-2OO-s/a和R1-6OO-s/a所有可能的自身异构反应渠道及对应的ΔE0K≠如图4所示,反应类型共可分为3类:(1) —OO—基团夺取—CH—或—CClH—的H生成—OOH;(2) —OO—基团进攻—CH—的C生成双环中间体;(3) —OO—基团夺取—CClH的Cl生成—OOCl(未能找到生成R1-2OOCl-s的TS,参考生成R1-6OOCl-s的能垒很高,认为未找到的TS不影响结论)。由图4可知,R1-2OO-s、R1-2OO-a和R1-6OO-s、R1-6OO-a经最佳自身异构渠道分别生R1-21OO-s、R1-2OOH12-a、R1-64OO-s和R1-61OO-a,需要克服30.9、26.7、23.2和24.7 kcal·mol-1的反应能垒,对应的高压极限速率常数分别是1.7×10-11、6.9×10-4、9.6×10-6和1.5×10-6s-1(温度为298 K),远低于它们在高浓度NO条件下与NO及HO2·的伪一级反应速率常数kNO[NO]=1.4 s-1和kHO2[HO2·]=0.02 s-1(基于[NO]=1.3×1011molecules·cm-3,[HO2·]=1.3×109molecules·cm-3,kNO=1.2×10-11cm3·molecule-1·s-1,kHO2=1.7×10-11cm3·molecule-1·s-1,温度为298 K)[3,47-49]。因此,在高浓度NO条件下,R1-2OO-s/a和R1-6OO-s/a主要与大气中的NO发生双分子反应生成烷氧自由基(RO·) (R1-2O-s/a和R1-6O-s/a)和有机硝酸酯(C10H8ClONO2)。而在低浓度NO条件下,过氧自由基(R1-2OO-s/a和R1-6OO-s/a)与NO及HO2·的伪一级反应速率常数为kNO[NO]=0.001 s-1和kHO2[HO2·]=0.02 s-1(基于[NO]=1.3×108molecules·cm-3,[HO2·]=1.3×109molecules·cm-3,温度为298 K)[3]。因此,在低浓度NO条件下,R1-2OO-s/a和R1-6OO-s/a主要与HO2·反应形成有机氢过氧化物(QOOH)和RO·。因此,生成的RO2·除转化生成闭壳层产物有机氢过氧化物和有机硝酸酯外,还会生成RO·,接下来主要讨论RO·中间体R1-2O-s/a和R1-6O-s/a的反应。
2.4 R1-2O-s/a和R1-6O-s/a的反应
与其他自由基转化生成的RO·的反应类似,R1-2O-s/a和R1-6O-s/a也可以发生单分子自身异构及解离或进一步与大气中的O2反应[29,50]。所有考虑的R1-2O-s/a和R1-6O-s/a的单分子自身异构与解离及与O2反应的渠道及对应反应的ΔE0K≠如图5所示。可以看出,4种RO·存在3种单分子异构化反应类型:(1) RO·的O原子攻击不同的—CH—的C形成含O双环中间体;(2) C—C键断裂生成相应的醛;(3) H迁移和C—H键断裂,生成相应的酮。对比R1-2O-s/a和R1-6O-s/a所有单分子自身异构化与解离渠道反应ΔE0K≠可知,R1-2O-s/a和R1-6O-s/a经环化生成R1-21O-s/a和R1-61O-s/a双环中间体是最可行的自身异构与解离反应渠道,对应的反应速率常数分别为3.8×108、8.3×107、6.8×1011和2.7×1011s-1(298 K)。对于RO·和O2反应,O2能分别夺取R1-2O-s、R1-2O-a和R1-6O-s、R1-6O-a的C2和C6上的H原子生成酮,反应的伪一级反应速率常数分别是1.7×103、9.1×102、6.2×102和3.2×102s-1(双分子反应速率常数分别为3.5×10-16、1.9×10-16、1.3×10-16和6.5×10-17cm3·molecule-1·s-1及O2浓度为4.9×1018molecule·cm-3,温度为298 K),低于对应的最可行的自身异构与解离反应速率常数。因此,R1-2O-s/a和R1-6O-s/a的主要反应渠道是通过单分子环化形成相应的双环中间体R1-21O-s/a和R1-61O-s/a。
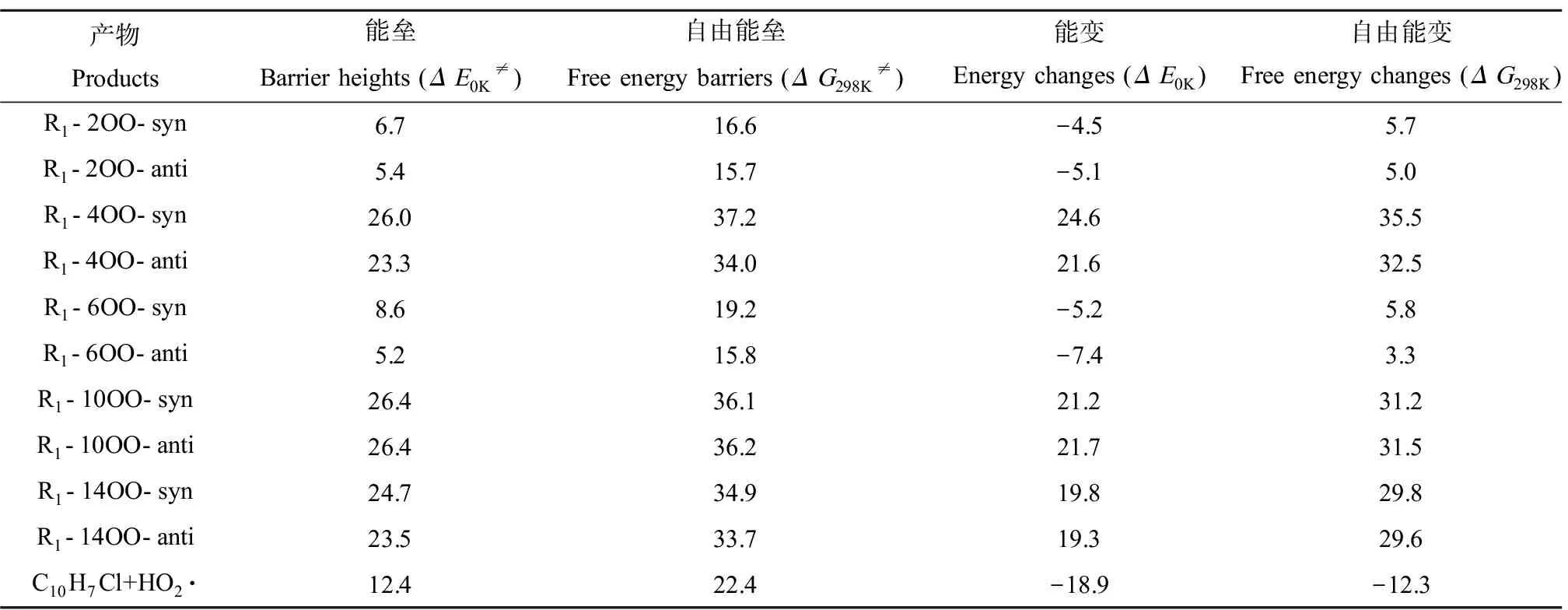
表2 在ωB97XD/6-311++G(3df,2pd)//ωB97XD/6-31+G(d,p)计算水平下,R1+O2反应的反应能和能垒Table 2 Reaction energy and barrier heights for reactions of R1 with O2 at the ωB97XD/6-311++G(3df,2pd)//ωB97XD/6-31+G(d,p) level (kcal·mol-1)

图4 在ωB97XD/6-311++G(3df,2pd)//ωB97XD/6-31+G(d,p)计算水平下,R1-2OO-s/a (a) 和R1-6OO-s/a (b)自身异构反应渠道和能垒(ΔE0K≠)(单位为kcal·mol-1)Fig. 4 Barrier heights (ΔE0K≠) for isomerization pathways of R1-2OO-s/a (a) and R1-6OO-s/a (b) at the ωB97XD/6-311++G(3df,2pd)//ωB97XD/6-31+G(d,p) level (all in kcal·mol-1)
3 环境意义(Implications)
本研究基于量子计算化学和过渡态理论,首次揭示了·Cl引发Nap的大气转化机制和动力学。计算得到的·Cl+Nap的主要的反应路径如图6所示。可以看出,引发反应主要为·Cl加成到Nap的C5位置生成·C10H8Cl。加成中间体·C10H8Cl后续主要与O2通过syns和anti这2个方向加成到C2和C6位置生成4种RO2·中间体R1-2OO-s/a和R1-6OO-s/a。在低NO浓度条件下,RO2·主要和HO2·反应生成氢过氧化合物(QOOH)和烷氧自由基(RO·);在高NO浓度条件下,RO2·将主要与NO反应生成RO·(R1-2O-s/a和R1-6O-s/a)和有机硝酸酯(C10H8ClNO3)。重要的是,使用ECOSAR模型预测生成的有机氢过氧化合物、有机硝酸酯的水生毒性强于其母体化合物Nap(有机氢过氧化合物、有机硝酸酯和Nap对鱼的96 h半致死浓度(LC50)分别为0.92、1.54和9.39 mg·L-1,对绿藻的96 h半效应浓度EC50为0.42、1.95和6.91 mg·L-1),表明·Cl引发Nap转化增加了Nap释放的环境风险[36]。生成的4种RO·主要是通过环化生成相应的双环产物R1-21O-s/a和R1-61O-s/a。因此,为进一步完善·Cl引发Nap的大气转化机制,将来需要研究双环中间体的后续转化机制和动力学;同时,为准确评估·Cl引发Nap转化导致的环境风险,将来有必要对转化过程中生成的闭壳层产物的毒性展开研究。本文的研究结果为全面评估·Cl引发萘的大气转化及环境风险提供了数据支持。
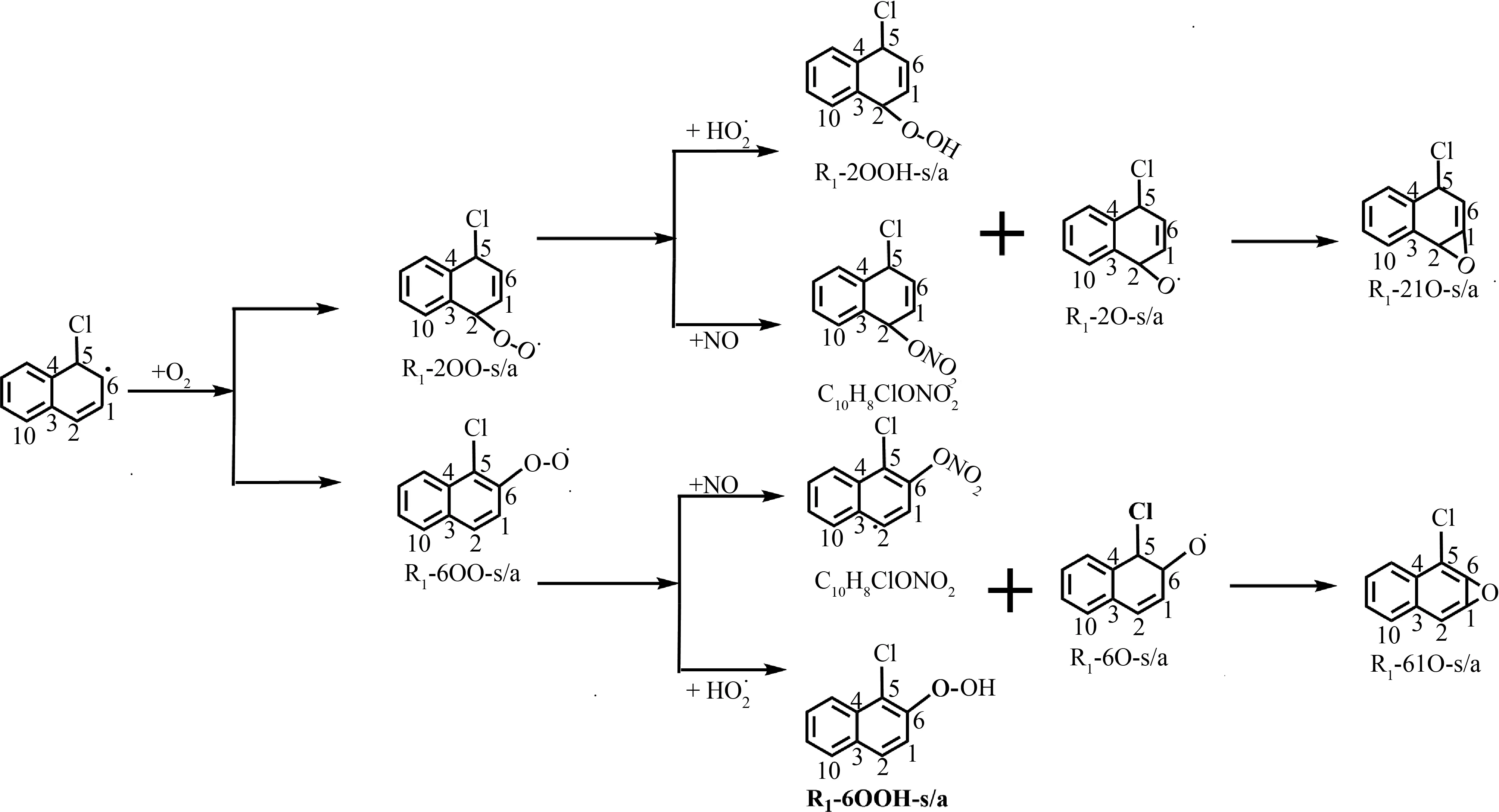
图6 ·Cl+Nap的主要反应路径Fig. 6 Main pathways for the ·Cl initiated reaction of Nap
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
数学年刊A辑(中文版)(2021年1期)2021-06-09
汕头大学学报(自然科学版)(2020年4期)2020-12-14
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
科学中国人(2018年8期)2018-07-23
中成药(2017年5期)2017-06-13
新高考·高一物理(2016年3期)2016-05-18
股市动态分析(2015年12期)2015-09-10
医学研究杂志(2015年12期)2015-06-10
