
聚羧酸减水剂活性大单体聚合机理及合成研究进展
2022-01-07 14:09张跃伟季泽尧宋春民施劲松娄大伟
吉林化工学院学报 2021年11期
张跃伟,季泽尧,祝 波,张 浩,宋春民,施劲松,汪 锐,付 强,娄大伟,*
(1.吉林化工学院 化学与制药工程学院,吉林 吉林 132022;2.吉林瑞吉特殊化学品有限公司,吉林 吉林132022;3.吉林化工学院 石油化工学院,吉林 吉林 132022)
中国作为全球最大的混凝土消费市场,混凝土减水剂的优化对于市场的发展尤为重要.在广泛使用的减水剂中,聚羧酸减水剂在各个工程指标如减水率、掺量、体积稳定性、保坍性、生产成本和环境友好性等方面相比传统的减水剂表现都较为良好[1],因此聚羧酸减水剂受到国内外混凝土行业的极大重视.聚羧酸减水剂(PCE)[2]因其易调节的分子结构被认为是最重要的混凝土外加剂之一,可适应于不同用途的混凝土,目前已在桥梁、铁路、水电大坝和地标性建筑等国家重大工程中得到广泛应用.在工程应用过程中,研究者和生产技术人员发现PCE对附着在混凝土骨料周围的黏土具有很强的敏感性[3],其工作性能受黏土矿物的不良影响.然而随着建筑行业快速发展,优质矿物原料日益减少,含泥骨料用量日益增多,这必将成为限制PCE大量推广使用的重要因素之一[4].
同时PCE的聚合单体价格居高不下,聚合单体的生产过程中,双键留存率不稳定,因此对单端不饱和长链烷基醚的生产工艺改良迫在眉睫.随着混凝土行业市场的不断扩大,专家预测,近5年中国聚羧酸减水剂市场将以超过4%的增长率持续增长.因此,对于聚羧酸减水剂(PCE)的活性大单体合成的深入研究至关重要.本文将对不同的高分子聚合机理、不同机理的催化剂和反应条件进行总结,结合目前单端不饱和聚乙二醇的生产和研究现状,对适合于工业化生产的工艺路线的研究进展和发展趋势进行展望,为持续增长的聚羧酸减水剂市场需求提供借鉴.
1 聚合机理
1.1 阴离子聚合机理
遵循阴离子聚合机理,环氧化物可在碱金属氢氧化物的作用下被醇盐进攻而发生聚合,由此可生产端羟基聚醚[5-8].工业上遵循此机理进行生产时,常用的催化体系多由单一碱金属的强碱性化合物构成,如叔丁醇钾、氨基钾及萘钾等有机强碱和以氢氧化钠为代表的无机强碱等.这类催化剂参与的聚合反应多为先由强碱与底物反应生成阴离子作为活性种,而后通过活性种进攻聚合单体来实现聚合完成的分步反应的过程.
合成的主体反应为环氧烷烃的开环聚合反应.聚乙二醇(Polyethylene glycol,PEG)的聚醚结构中,醚键由于其富电子特性,在Lewis酸碱定义中属于碱性键,因此,通常而言,大多数环醚的聚合需要体系中存在阳离子引发剂才能进行开环聚合,但当环氧环过小时,被剧烈弯曲的碳氧键存在很大的张力.故分子结构中存在三元环的环氧化物反应活性很高,因此环氧乙烷等三元环氧化物不仅可在阳离子引发剂的催化下进行开环,而且在阴离子引发剂的作用下同样可以发生开环聚合获得聚醚[9-10].在环氧化物的阴离子聚合机理中,常见的阴离子催化剂多为由碱金属构成的强碱性化合物,如叔丁醇钾、氨基钾及萘钾等有机强碱和以氢氧化钠为代表的无机强碱等.此外,任何可为反应体系提供活泼氢的结构如水、醇等均可作为引发剂.在阴离子催化剂催化的聚合反应中,链引发阶段时引发剂进攻环氧化物的2位亚甲基,引发环氧化物的开环,得到醇盐阴离子,醇盐阴离子在链增长阶段再一次进攻环氧化物单体,使得环氧化物继续开环并使其链接到醇盐阴离子上,醇盐阴离子的聚合数得到增长,实现链式反应[11].如用醇盐阴离子RO-引发环氧乙烷遵循阴离子聚合机理开环聚合的反应示意如图1所示.

图1 醇盐阴离子RO-引发环氧乙烷开环聚合反应示意图
在环氧乙烷阴离子聚合过程中,链引发、链增长和质子交换反应的相对速率不同,导致了其不同的聚合结果:
(1)快引发、慢增长、无质子交换,得到相对分子质量分布窄的聚合产物;
(2)慢引发、快增长,则会得到在不同时间内生成的聚合物链,最终使得聚合产物的相对分子质量分布变宽.
反应体系中聚合物醚醇与聚合起始剂的相对酸度决定了分子间H+的交换速率.当二者的相对酸度较为接近时,质子交换将贯穿于整个聚合反应中,影响最终聚合度,并使得聚合物的PDI值增大;当起始剂的相对酸度远高于聚合物时,此时反应方程式中的平衡点将向右移动,由此反应存在首先将体系中所有的起始剂活化后再进行链增长的趋势,这种趋势使得到的聚合产物的相对分子质量分布较起始剂与聚合物的相对酸度差异不大时更窄[12].
1.2 阳离子聚合机理
当聚合反应遵循阳离子开环聚合机理进行时,由于与阴离子聚合机理的差异,一般选取含氢酸作催化体系的效果最好,并在进行聚合机理和反应动力学的研究时优先选用.同时用于环醚进行阳离子开环聚合的催化剂主要分为:含氢酸[13]和Lewis酸[13-14]两大类.
在进行阳离子聚合的过程中,含氢酸提供的H质子首先进攻三元环氧化物氧桥上的氧原子,使得被进攻的三元环氧化物形成类似鎓盐的过渡态结构,但形成的中间产物极不稳定,会快速进行开环形成活性阳离子增长种,而后进攻聚合底物并发生分子链的延长而使分子量快速增加.由于该聚合反应的开环步骤为反应的限速步,且原料酸可以很快转化为加成物,因此产物的分子量分布较窄.通过实验可知,当聚合度较低时,聚合产物的分子量分布曲线遵守Poisson分布曲线或Florry分布曲线[15].但此种催化体系仍存在副反应多难以获得高纯的产品,聚合产物的相对分子量均较低无法生产高分子量产品等缺点,所以该机理仅用于工业化生产聚四氢呋喃[16].
从绝对意义上说,阳离子聚合机理归于连锁聚合,因为动力学链的承担者(活性增长种)在反应过程中是阳离子.聚合物增长链的端基由这些阳离子活性种组成,并且这些阳离子活性增长种在不同的反应体系中为不同的存在形式.在开环聚合反应中,阳离子活性增长种多以“鎓离子”形式存在于反应体系中[17],以THF在HOCH2CH2OH的存在下,由BF3·Et2O催化阳离子开环聚合反应为例[18],反应示意图如图2所示.

图2 三氟化硼乙醚催化四氢呋喃开环聚合反应示意图
碳正离子的反应活性,即使“稳定”,也是很高的.这导致了影响阳离子聚合发展的至少三方面的问题:
(1)即使在低温条件下,链增长速率也非常高,使得反应难以控制;
(2)分子异构化及不同种类的链转移时常发生,使得难以获得纯净的产物;
(3)由于聚合反应对杂质十分敏感,故对原料的要求较高.
与环氧化合物的阴离子开环聚合不同,环氧化物的阳离子开环聚合一般难以自发终止,故需加入终止剂使聚合终止,反应方程式中的XA代表终止剂,常用的终止剂如水、醇、酸等.同时由于阳离子开环聚合的限速步为链引发,因此只要链引发步骤发生,聚合单体就能与活性增长种迅速结合,所以经阳离子聚合机理得到的聚醚的分子量分布很窄.但获得的产物分子量不高,同时因存在较多的副反应,产物的纯度也较低[19].
1.3 配位聚合机理
利用配位聚合机理进行环氧烷烃配位聚合反应可使用的催化体系类型很多,大多数的配位催化剂为锌、铝、铁和钴的配合物.同时生产中绝大部分配位催化剂不能单独催化反应,而是通过与其他组分混合成为多组分复合体系来发挥催化作用,目前配位聚合催化剂研究中最活跃的方向是双金属氰化物络合物(Double Metal Cyanide,DMC)的制备及其催化环氧烷烃开环聚合生产聚合物中的应用.配位聚合催化剂相较于传统碱金属的碱性化合物如KOH等催化剂具有可生产高分子量的聚醚、产物的分子量分布较窄及反应原料的反应程度较完全等优点[20],因此其在聚醚生产理论研究和工业应用中的地位非常重要.
以DMC催化环氧乙烷聚合为例说明配位聚合机理,如图3所示.
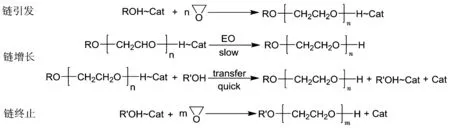
图3 DMC催化环氧乙烷聚合示意图
通过对聚合机理的分析研究,发现锌-钴双金属氰化络合物催化环氧烷烃进行聚合的反应属于活性聚合机理,该催化剂的活性中心为锌原子,活性中心周围存在5个提供配位氧原子的氢氧根离子与其进行络合,同时聚合物的分子量仅由聚合底物与引发剂的投料比控制.
在遵循配位聚合机理的聚合反应中,双金属氰化物络合物(DMC)在环氧烷烃的作用下被激活,催化过渡态的活性结构由起始结构与DMC中锌原子络合形成,而后起始结构通过锌原子转移到聚合单体上,使得起始结构的碳链得到增长.同时起始结构与催化活性中心之间存在基团的交换,且基团互换的速率相较于起始结构所产生的聚合活性种的增长更快.因为该特征,使得分子量相差不大的聚醚多元醇的合成成为可能,即当所有起始物分子的聚合过程同时启动时,所得产品的分子量便相差不大.若要实现本目标,仅需使反应过程中的链转移步骤进行时,在活性中心与起始结构之间转移的基团不发生异构化即可.
2 研究与应用
2.1 阴离子催化研究进展
利用阴离子作为聚合反应的活性增长种时,反应仅存在链引发和链增长两个步骤.随着活性分子碳链的增长,阴离子活性增长种的反应活性逐步下降,对聚合底物的进攻能力逐渐减弱,使得聚合反应趋于终止.同时由于聚合过程中每个阴离子活性增长种的反应活性不同,获得产物存在分子量不高、分子量分布宽等缺点.但利用阴离子开环聚合机理进行工业生产时,相较于其他的催化体系所使用的昂贵催化剂,阴离子聚合的催化剂较为易得,且工业生产对设备的要求不高,因此目前是工业生产聚合物的主流方案[21-24].
为了克服阴离子催化剂存在的缺点,研究人员对阴离子催化剂进行了较多的研究,对阴离子催化剂的改进取得了一定的效果.如Perry等[25]用实验证实了环氧化物的阴离子聚合反应遵循逐步活性聚合机理,通过进一步研究,他们发现低分子量的聚合物也可作为聚合反应的起始剂使用,低聚合度的聚乙二醇在叔丁醇钾的作用下产生的聚乙二醇钾可进攻环氧乙烷并与其发生链增长,在链增长完成后,若继续加入环氧乙烷,则聚合反应将继续进行,聚合物的分子量也将增大.利用阴离子聚合反应的这个性质,Holland[26]利用分批加入催化剂及环氧乙烷避免了一次性加入聚合单体过多而使聚合反应速率减慢的可能,同时得到了高聚合度的聚环氧乙烷(PEO),表1为阴离子催化剂研究实例.

表1 阴离子催化剂研究实例
此外,聚合过程中聚合单体的异构也是导致聚合物不饱和度升高的重要因素.Becker和Wagner研究发现[27],冠醚(18-Crown-6)因其特殊的结构可抑制聚合过程中聚合单体的异构化,使得产物的不饱和度下降.同时由于冠醚的辅助作用,聚合反应的速率也将得到提升.Gonod等[27]通过设计正交实验探究了产物的不饱和度与反应体系中加入冠醚的量和反应温度之间的关系,他们发现在较宽的反应温度范围下,若加入冠醚的量保持不变,则产物的不饱和度相对稳定.同时加入的冠醚相对越多,所得产物的不饱和度便越低,但最小达到初始水平的三分之一左右后便不再减小.并根据筛选出的反应条件制得了更大分子量的聚醚.
2.2 阳离子催化研究进展
20世纪50年代时,人们开始研究环氧化物的阳离子开环聚合机理,Eastnam等人[28]利用三氯化铝和三氟化硼乙醚作为催化剂,实现了环氧乙烷的阳离子聚合,但得到的产物纯度相较于阴离子聚合较低,且无法得到高聚合度的产品.Grinerich等人[28]研究发现遵循此机理的聚合反应的聚合度存在一个最大值,分子量达到极值后不再继续增长.后来,为获得更大分子量的聚合产物,英国的Sterart[29]通过水合三氟化硼作为催化剂,使得阳离子聚合的分子量达到了更高的范围,使得阳离子聚合机理具有了工业化的可能.目前,聚环氧氯丙烷、聚四氢呋喃及环氧丙烷-四氢呋喃共聚醚等[30]产品大多采用阳离子聚合生产,其中产量最大的为BASF公司[30]的聚四氢呋喃,是阳离子聚合工业化生产最重要的成果,表2为阴离子催化剂研究实例.
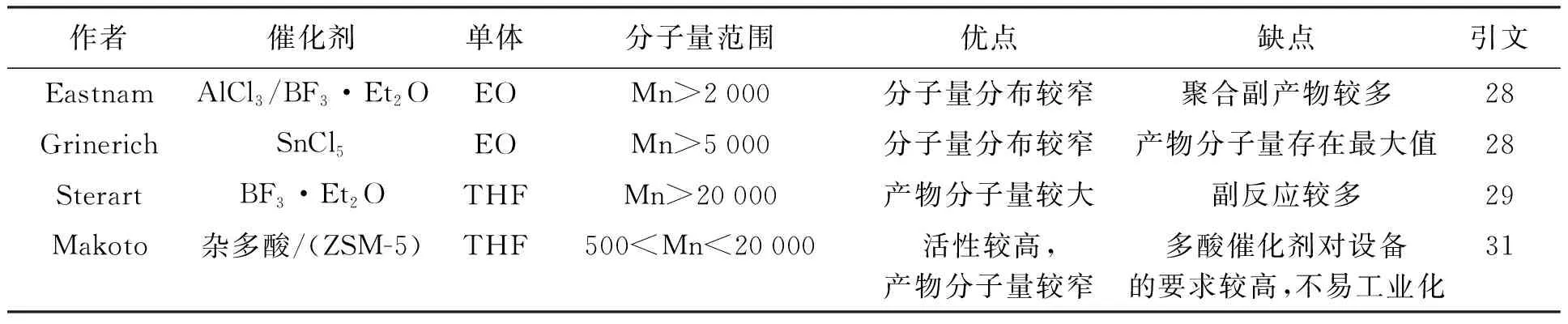
表2 阳离子催化剂研究实例
从20世纪80年代开始,以固体酸(杂多酸)作为新型催化剂[31]引起了学术界的重视,并成功制备出了几种可以进行工业化生产的催化剂,随着对杂多酸催化体系研究的深入,描述杂多酸催化机理的杂多酸标准液相模型也被提出.固体酸由于本身为固体,储存较液态酸更容易,同时因为其固态的结构使得催化的活性中心较液体催化剂更加集中,故催化活性更好.同时催化活性中心的集中也使得其在用量更少的同时,催化聚合获得的产物分子量分布更集中.虽然固体酸催化剂的优点很明显,但固体酸催化聚合无法避免聚合单体在反应过程中的异构化,导致产物的纯度不高,同时由于催化剂为固态,无法通过常规的过滤等分离手段与产品进行分离,导致生产的后续分离成本较高.同时对生产设备的要求较高也限制了该催化体系在工业上的推广使用[31].
2.3 配位聚合催化剂研究进展
常见的阴离子聚合催化体系和阳离子聚合催化体系都存在不适于工业化生产的缺点,为提高工业化生产的效率,人们开发了以中等亲核性金属原子为配位中心的一系列用于催化环醚聚合的配合物.如含有镉、钴、铁等元素的配合物都具有良好的催化活性.此外,部分配位基团中若存在稀土元素可使催化效率得到提高.但目前所开发的配位催化剂单独使用的效果均不理想,但若反应体系中存在活泼氢,可使配位基团得到活化,显著改善配位催化剂的催化活性.按照催化体系中配位基团组分的不同,大致有下述几种分类.
2.3.1 Fe(Ⅲ)催化剂
最先研究的是以三价铁为配位中心的配合物,主要用于氧化烯烃的聚合.1955年,Baggett等[32]首次报道了利用Fe3+系配位催化剂催化环氧烷烃开环聚合.根据催化体系中提供活泼氢的物质的不同,Fe3+系的配位催化体系可分为三氯化铁-磷酸体系及三烷氧基铁-水体系两类,在前人的基础上,Colcough等[32]分别对两种不同催化体系的配位催化活性中心的构造进行了研究,认为该活性中心为配位铁原子部分水解后的络合物.
后来的实验证实了三价铁催化聚合的反应机理,即配位铁原子首先将反应体系中游离的活泼氢转移至环氧化物的氧原子上,形成不稳定的中间过渡态结构,而后重复此步骤形成低聚合度的链节后,再按配位机理发生聚合[32].即反应体系中先产生低聚合度的引物,而后再进行聚合,因而可获得高分子量(数万以上)的产物.
2.3.2 以烷基金属为基础的催化剂
烷基金属催化剂因其烷基配位导致配位金属原子周围的空间位阻很大,导致其催化活性下降,但当反应体系中存在其他基团使得配位金属原子暴露出来时,其催化活性将获得极大改善.故使用此类催化剂时一般不单独使用,应与其他能与其发生螯合的物质合并使用.常见的烷基金属催化剂有三乙基铝、异丁基铝、二乙基锌、二乙基镁等.烷基金属催化体系主要分为两种:金属卟啉体系和烷基铝、烷基锌体系.
(1)金属卟啉络合体系催化剂
20世纪70年代末,人们研究发现金属卟啉配合体系[33]克服了传统烷基配位金属化合物催化活性中心的空间位阻问题,具有良好的催化作用.催化剂构成主要为配位金属离子、亲核性富电子基团及有机配体.
金属卟啉配合体系所合成的高聚物的分子量分布较窄,但存在聚合速率慢的缺点.为改善其反应速率,研究人员将Lewis酸与卟啉体系相结合,使得金属卟啉配合体系催化的聚合反应速率显著加快.
(2)烷基铝、烷基锌体系催化剂
该催化体系主要由烷基铝和一些改性组分构成,改性组分主要包括水、醇、酸及胺等.研究者开发出了烷基铝-水体系和烷基铝-磷酸催化体系,通过改性组分对配位金属原子周围空间位阻的改善,这两种催化体系相较于单独使用烷基铝进行催化显著提高了催化活性.同时,Furukawa等[34]也提出了以锌原子代替铝原子作为配位金属原子的催化体系,扩展了以烷基金属为基础的催化剂类别.
通过本催化体系制得的聚合产物往往分子量分布不够集中,这显示了催化缔合体活性中心并不唯一[35].故无论是从技术还是成本的角度,这两种催化体系均不适用于工业生产.
2.3.3 烷氧基铝催化剂
Robert和Kayan等[36-37]发现,与烷基铝等烷基金属催化剂的作用机理不同,烷氧基铝可与聚合底物形成环状过渡态,通过脱去一分子醇来使得聚合反应能够发生.在特定的反应条件下,烷氧基铝类配合物表现出极高的催化效率,同时有多条配合物的分子链连接在配位金属原子上.随着反应的进行,反应体系中醇的比例逐渐上升,此时醇和烷氧基铝分子数的总和即为发生聚合的聚合单体数目,且产物的相对分子量较为接近,同时具有制备带官能团的聚醚产物的可能.烷氧基铝催化剂催化生产的聚合物中仍然含有部分的双键端基.一种可能的机理解释如图4所示.

图4 烷氧基铝催化剂可能机理示意图
2.3.4 双金属氰化物催化剂
DMC催化获得的产物具有较窄的分子量分布及较高的饱和度等优点,同时还可生产相对分子量较大的产品,这是传统开环聚合的催化剂所不具有的优势[38].Generaltire& Rubber公司多年的研究与应用表明,Zn3[Co(CN)6]2和Zn3[Fe(CN)6]2是催化效率最高,各项指标最优秀的两种催化剂[36-38].
DMC催化剂具有如下特点[39]:
(1)催化剂的催化性能可能存在很大差异;
(2)催化聚合时聚合底物的活化期更长,生产的能耗更大,同时因为聚合过程剧烈放热,反应体系的热效应难以控制;
(3)绝大部分催化剂必须使用聚醚低聚物充当起始剂,低聚合度的大分子无法作为聚合反应的起始剂;
(4)DMC催化体系催化的聚合产物需进行二次处理,生产成本较其他催化体系更高;
DMC催化生产的聚醚产品的相对分子质量分布较窄,同时产品的羟基官能度下降不多.对这个现象目前存在一种解释,即认为体系聚合过程中的端基重排被催化活性中心消除.催化剂的分离是利用DMC催化剂生产聚合产物过程中最大的难题.工业上常用的分离手段难以从产物中除去DMC催化剂,增加了使用DMC催化剂进行工业生产的成本.此外DMC催化剂相较于其他常用的催化剂毒性更大、价格更高,也限制了DMC催化剂的推广使用.
2.3.5 磷腈(PZN,Phosphazene)类催化剂
日本三井化学株式会社的研究人员[40]认为催化活性中心上分布的阳离子电荷在聚合底物与活性中心之间的转移是聚合反应能够进行的关键,故而要提高催化剂的催化效率应加快活性中心与聚合底物之间的电荷有效转移.同时提出了作为环氧化物开环聚合的催化剂应满足以下几点要求:
(1)不含金属元素;
(2)催化结构的尺寸应足够大;
(3)分子中阳离子的电荷应分散在整个催化活性中心;
(4)为避免杂质的产生,活性位点应仅有一个.
此外,分子结构对称可使分子的稳定性提高,提高催化剂的使用寿命.
符合上述要求且分子结构对称的催化体系主要为结构中存在强碱性的磷氮(P=N)双键磷腈类催化剂[41],但目前上市的磷腈类催化剂的生产成本较高,限制了该类催化剂的使用.同时由于其分子内磷氮双键的存在,使得该化合物能够与弱酸反应,如空气中存在的二氧化碳,这同时也限制了该类催化剂的推广使用.几种常见催化剂性能对比如表3所示.

表3 几种常见催化剂的性能对比
3 结 论
结合研究现状而言,阴离子催化剂因其较为温和的反应条件和相对稳定的产率仍作为高分子聚醚的主要生产方式;阳离子聚合因其副反应过多不易提纯和对设备的要求较高而仅适用于聚四氢呋喃的工业化生产;配位聚合作为新兴的研究方向,工业化的实例不多,但配位聚合催化剂的反应条件和产物的分子量分布都较好,开发前景十分广阔.
就工业生产现状而言,聚羧酸减水剂大单体的生产还是以阴离子反应机理为主,虽然使用阴离子机理进行生产会导致双键的保留率下降,但其具有良好的生产成本控制和可生产较大分子量的优点,使得双键保留率下降这一缺陷也可以被接受.对于阴离子聚合的催化剂研究可以向能在较高温度下仍旧能够做到较高的双键保留率的方向努力;可以尝试先将不饱和的起始物先保护起来,待聚合完成后再脱保护得到不饱和的聚合大单体的生产路线.对于阳离子聚合催化剂而言,未来的研究方向应是开发如何使得聚合物的分子量突破所谓的“聚合最大分子量”并且使得聚合反应可控;由此可以尝试降低催化剂的催化活性,将现有的催化剂钝化,使得聚合反应的速率下降进而使得反应可控.配位聚合催化剂的开发方向较多,且不同系列的催化剂各有优劣,未来的研究方向可以尝试在保持本系列催化剂的性能的条件下,使其具有部分其他系列的催化剂的特性,进而开发出性能更好、价格更低廉、更适合工业化生产的催化剂.
猜你喜欢
燃料化学学报(2022年5期)2022-05-30
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
当代陕西(2019年6期)2019-04-17
合成化学(2015年4期)2016-01-17
海军航空大学学报(2015年1期)2015-11-11
现代农业(2015年3期)2015-02-28
应用化工(2014年1期)2014-08-16
无机化学学报(2014年4期)2014-02-28
郑州大学学报(理学版)(2013年2期)2013-03-11
郑州大学学报(理学版)(2012年4期)2012-03-25
