
[Ir(ppy)2(X-ppz)]+配合物结构和光谱性质的量子理论研究
2022-01-07 14:09聂建航王子轩贲虹博张建坡
吉林化工学院学报 2021年11期
聂建航,王子轩,贲虹博,张建坡,金 丽
(吉林化工学院 化学与制药工程学院,吉林 吉林 132022)
近几年,由于金属铱配合物具有独特的光物理特性,引起了材料学家的广泛关注[1-3].传统的铱配合物在有机发光二极管(OLED)领域中有三大优势:首先其具有相当长的激发态寿命,可达到几十微秒,从而消除短寿命的自发荧光.其次它具有大的斯托克斯频移,这可以最小化其自猝灭过程,因此铱配合物显示出覆盖整个可见光谱的可调激发和发射波长.最后它可以有效地阻止固态聚集的形成.例如,在2012年,So集团开发了一种新的铱配合物,含有苯基吡啶配体和一个庞大的三甲基甲硅烷基二甲苯基团,在532 nm处发出黄绿色光.铱配合物作为掺杂剂在其中表现出高的器件性能,在掺杂剂浓度为6wt%时,EQE为12.7wt%,LE为45.7 cd/A,其有效地阻止了固态聚集的形成[4].
最近一类环金属化铱(Ⅲ)配合物[Ir(ppy)2(L)]+[ppy=2-苯基吡啶;L=双(吡唑-1-基)甲烷或双(3,5-二甲基吡唑-1-基)甲烷]被合成[5],并研究了它们的电化学和光物理性质.与常用的含N^N辅助配体的衍生物如联吡啶或邻菲罗啉相比,这些含非π-电子共轭辅助螯合物的配合物表现出明显的蓝移发射.为进一步揭示铱配合物的发光规律,本实验选取一类混合配体[Ir(ppy)2(X-ppz)]+[ppy=苯基吡啶,ppz=2-吡啶基吡唑,X=氢(1),X=苯基(2和3连接位置有差异)]配合物采用量子计算方法进行理论探究,考查配体的差异对分子结构和发光性质的影响.
1 计算方法
运用密度泛函中的B3LYP[6]和CIS (单激发组态相互作用方法)[7],对分子1-3的基态和激发态进行详细的理论研究.分子1-3优化过程中均无对称性约束,计算中,对Ir原子使用赝势LANL2dz基组[8],对C、N、H原子使用6-31G*基组.以基态和激发态结构为基础,根据电子垂直跃迁原理,使用TD-DFT[9]方法在充分考虑溶剂介质 (PCM介质模型)[10]影响的基础上得到3个分子在二氯甲烷溶剂中的光谱特征.所有计算使用Gaussian09程序包在高性能曙光机架式服务器上完成.
2 结果和讨论
2.1 基态和激发态结构
3个分子的基态结构优化图和代表性的原子编号如图1所示.相对应的结构参数和分子1所对应的实验值列于表1.其中,基态时配合物1的Ir-N1(2.216Å)、Ir-N3(2.171Å)、Ir-N4(2.080Å)分别与相应实验值相差0.079Å、0.045Å、0.034Å,在考虑计算环境与真实分子环境的差异情况下,说明计算结果合理.

图1 分子基态结构优化图
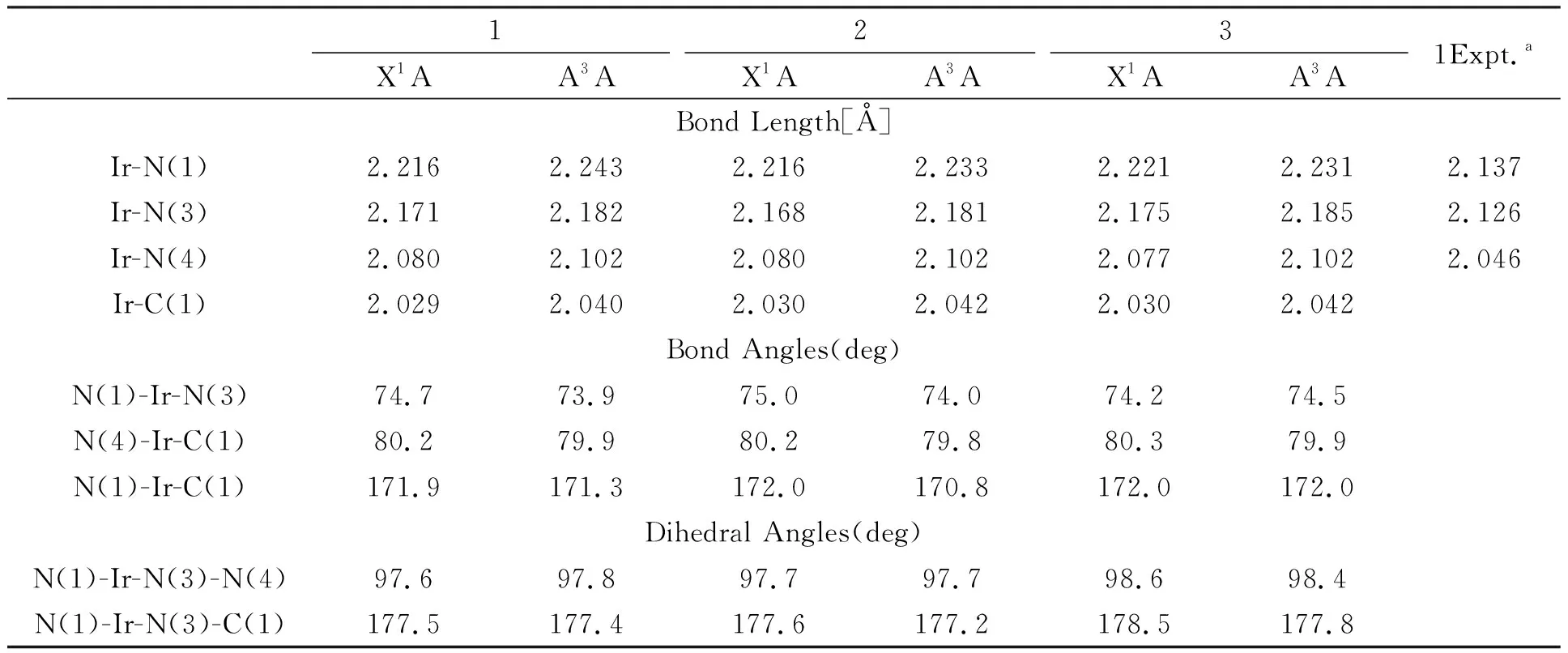
表1 分子基态(S0)和激发态(T1)主要结构参数和分子1的实验值
对比3个分子的主要结构数据,可看出给电子基团苯环的引入并没有使配合物的主体结构发生较大改变,只是苯环位置改变时使ppz配体发生轻微扭曲.其中,N1-Ir-C1键角值为173.5°,说明N、Ir、C 3个原子近似于在一条直线.二面角N1-Ir-N3-N4计算值为97.6°,说明ppy和ppz配体接近垂直.
激发态时, Ir-N(1)、Ir-N(3)、Ir-N(4)、Ir-C(1)键均变长.这是由于激发态时金属的电子云向配体转移,在增强配体本身的电子云排布的同时,削弱了金属和配体的相互作用,对应着最低能吸收的MLCT跃迁[12]本质.而键角和二面角的变化有增大、有减小,但变化幅度都不大,显示出电子的激发,对分子主体结构影响并不明显.
2.2 配合物的吸收光谱
以基态优化结构为基础,运用量子化学中的TD-DFT方法得到了3个分子在二氯甲烷溶剂中的吸收光谱.将3个分子的典型吸收光谱数据(跃迁轨道、波长、能量、震荡强度、跃迁性质)列于表2中.并把其涉及的分子轨道成分展示在表3中.配合物1-3的Guassian型吸收光谱曲线在图2中展示.

表2 3个分子在二氯甲烷液体中的光谱数据及实验值
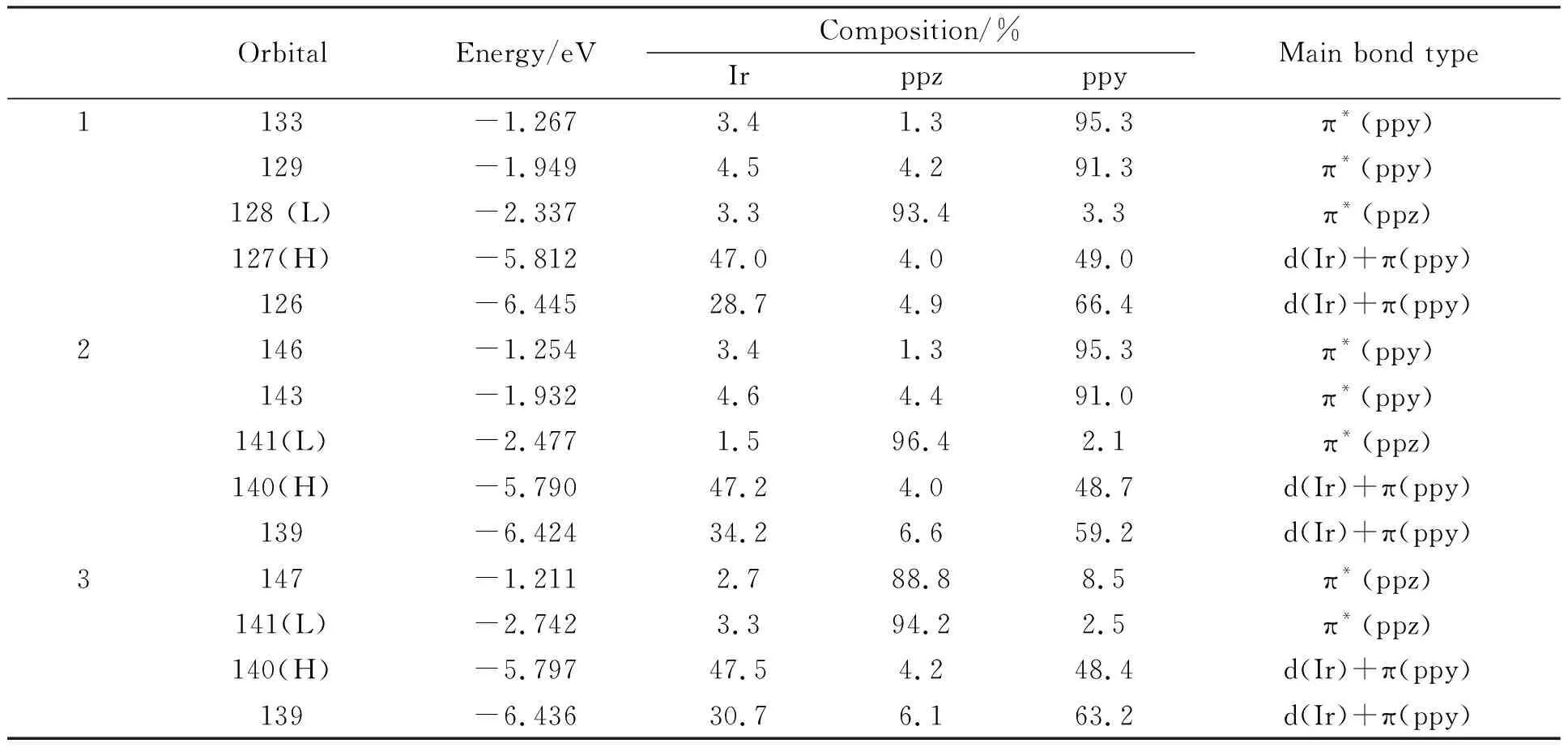
表3 3个分子在二氯甲烷液体中的电子吸收和跃迁所对应的轨道相关成分和能量
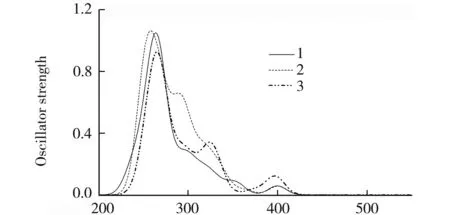
λ/nm图2 分子在二氯甲烷溶剂中的吸收曲线(模拟图)
来自表2和图2中的相关数据,3个分子均有两个高强度吸收带和一个弱吸收带.3个分子最低能吸收位于447.91(2.77)、456.52(2.72)、516.90(2.40)nm,轨道127→128的激发对分子1在447.91 nm吸收起主要贡献,轨道127为最高占据分子轨道,由47.0%的金属Ir和49.0%的ppy配体的π成键轨道构成,与此相对轨道128为最低空轨道,由93.4%的π*(ppz)配体占据.因此,该跃迁是属于d(Ir)+π(ppy)→π*(ppz)的金属到ppz配体和ppy配体到ppz配体之间的电荷转移(MLCT/LLCT)跃迁.同理,2和3位于456.52和516.90 nm的吸收也是来自HOMO到LUMO的跃迁,其区别在于苯环的加入,使最低空轨道上的ppz配体成分增加0.8%~3%.通过分析3个配合物的最低能吸收波长可以发现,萘环的引入(2)并没有造成波长的显著红移,与基本分子1相比只有8.61 nm的变化,但是随着苯环连接位置的变化(3)出现较显著的红移,与基本分子1相比有68.99 nm的变化.实验上也观测到分子1的吸收波长,其中最低吸收波长为411 nm,与计算值相差36.91 nm.
配合物1-3第1个明显吸收带与最低能吸收的跃迁强度相比有较大的提升,该位置跃迁波长集中在393~401 nm之间,波长相差较小.根据上文的分析,它们分别由HOMO→LUMO+1、HOMO→LUMO+2、HOMO-1→LUMO贡献.跃迁起始轨道都占据在Ir和ppy配体上,而终止轨道均落在ppy配体上,因此该跃迁具有与最低能吸收相区别的金属到ppy配体和ppy配体内部电荷转移(MLCT/ILCT)跃迁性质.
配合物1-3最明显的吸收峰可以从图2中看到,跃迁波长也非常相近,在260~270 nm之间.配合物1的268.99 nm处吸收峰属于分子轨道126→133的激发捐献,分子轨道126由28.7%的金属Ir和66.4%的ppy配体的π成键轨道占据,而轨道133则主要为π*(ppy)型轨道,该跃迁具有与第1个明显吸收带相似的跃迁性质.基于上面的分析,分子2在该位置的跃迁也拥有与1相似的跃迁性质,而配合物3的139轨道由30.7%的d(Ir)和63.2%的π*(ppy)配体占据,而147轨道主要由ppz配体占据,因此该跃迁被指认为区别于1和2的MLCT/LLCT的混合跃迁,ppz配体发挥了较大作用.
2.3 配合物的磷光发射光谱
3个分子在二氯甲烷中的发射光谱数据如表2所示,其T1态磷光发射位于512.02 nm(1)、501.14 nm(2)、562.29 nm(3).配合物2和3被指认为起源于HOMO→LUMO的激发,HOMO由金属Ir和ppy配体贡献,而LUMO轨道只由ppz配体捐献,因此最低能吸收具有相似的跃迁性质.但是分子1的发射捐献与分子2和3略有不同,它的最低能发射由分子轨道129→127贡献,并非是HOMO→LUMO,而是HOMO→LUMO+1.比较3个分子的吸收和最低能磷光发射(HOMO→LUMO/HOMO→LUMO+1),它们都具有类似的轨道成分和跃迁特性,其能量差值分别为0.35、0.25和0.19 eV.改变配体的结构和位置能使其边界分子轨道能发生明显改变,吸电子基团的加入使其发射波长红移,但吸电子基团位置的改变对发光颜色的影响更加明显.
3 结 论
从理论上详细研究了一系列[Ir(ppy)2(X-ppz)]+[ppy=苯基吡啶,ppz=2-吡啶基吡唑,X1=H,X2-3=苯基(2与3的区别在于苯环的连接位置不同)]配合物.研究结果显示:给电子基团苯基的加入对此类分子的主体结构影响不大,但是苯基连接位置改变可以导致ppz配体自身发生较明显的扭曲.3个分子的吸收和发射均具有相似MLCT/LLCT跃迁,它们的HOMO均由金属和苯基吡啶配体成分构成,而LUMO轨道主要占据在二联吡啶配体上.通过比较他们的分子轨道能级,发现引入取代基和改变配体连接位置都能使其边界轨道能发生有规律的变化.不同的给电子和吸电子基团均能使HOMO和LUMO轨道能发生明显改变,从而导致它们的跃迁波长发生蓝移或红移,本文吸收和发射波长均具有红移趋势.因此,可以通过改变不同的取代基团来改变此类配合物的发光颜色.
猜你喜欢
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
农药科学与管理(2019年8期)2019-11-23
原子与分子物理学报(2015年3期)2015-11-24
化学工业与工程(2015年1期)2015-02-10
读写算·教研版(2014年12期)2014-09-01
原子与分子物理学报(2014年1期)2014-03-20
