
高效液相色谱法测定保健食品中七烯甲萘醌
2021-12-31 07:30程岁寒覃先武朱金玲潘存锋张海波张彦
化学分析计量 2021年12期
程岁寒,覃先武,朱金玲,潘存锋,张海波,张彦
(酵母功能湖北省重点实验室,安琪纽特股份有限公司,湖北宜昌 443003)
维生素K2是一类含有2-甲基-1,4-萘醌母核结构的脂溶性维生素,主要由微生物代谢产生。维生素K2在改善血压、降低心脑血管疾病发生率以及预防骨质疏松等方面具有重要的药用价值。维生素K2又可表示为MK-n,n代表异戊二烯基团的数量。其中,七烯甲萘醌(MK-7)被认为是MK-n类物质中生物活性和生物利用度较优的成分之一。MK-7 在体内参与骨代谢,具有预防骨质疏松的功效,同时还能诱导肝癌、胃癌、肺癌、骨肉瘤等的癌细胞凋亡,具有广泛的抗癌功效[1-2]。日本、美国FDA 及欧盟先后批准MK-7 作为食品及强化食品添加剂使用,2016 年中国国家卫计委第8 号公告批准MK-7 作为食品营养强化剂可在调制乳粉中使用[3],并被国家市场监督管理总局于2016 年发布的205 号公告收录于保健食品原料目录,适用于所有人群,其每日允许摄入量为10~100 μg[4]。
近年来,MK-7 因其保健功效显著被广大企业及消费者接受,市场上含MK-7 的保健食品多以油脂为基质,而对于脂质本底高的样品,超声提取法往往会引入严重的杂质干扰。MK-7 检测方法报道较少,主要有高效液相色谱-紫外(HPLC-UC)法[5-12]、高效液相色谱-荧光(HPLC-FLD)法[13-15]、高效液相色谱-质谱联用(HPLC-MS)法[16-17]。2016 年国家卫计委第8 号公告中规定了原料中维生素MK-7含量的测定方法,样品用异丙醇超声溶解后用反相色谱法进行测定[3]。许多报道中也使用异丙醇、乙酸乙酯、正己烷等酯溶性溶剂对样品中的MK-7 进行提取,然后进行反相或正相色谱法测定[8-10]。此类方法操作简便,但不适用于高油脂食品。孔凡华等[13]将高脂样品经脂肪酶酶解、异丙醇分散后,用正己烷提取样品中的维生素K1和K2,采用高效液相色谱C18柱将维生素K1和K2与其它杂质分离,再以锌柱柱后还原,使非荧光化合物维生素K1和K2产生荧光,再经荧光检测器检测,以外标法定量,但此类方法样品处理需要进行萃取、浓缩、复溶等步骤,操作复杂,耗时较长,易造成维生素K 损失,且需要高效液相色谱仪配置柱后衍生器,方法对人员及仪器要求较高。而HPLC-MS 设备成本较高,暂不易于推广。
笔者拟建立保健食品中MK-7 的高效液相色谱测定方法,含油脂类食品经脂肪酶水解以除去油脂的干扰,样品中的MK-7 使用异丙醇提取后,经C18型色谱柱分离,采用紫外检测器测定,减少了萃取、复溶等操作。本方法操作便捷、准确、快速,为保健食品中MK-7 的检测和质量控制提供了技术支持。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Waters e2695 型,配有2998 PDA 检测器和Empower 色谱工作站,美国沃特世公司。
电子恒温水浴锅:DZKW-4 型,北京中兴伟业仪器有限公司。
电子天平:XS105 型,感量为0.01 mg,瑞士梅特勒-托利多公司。
离心机:3-15 型,美国西格玛公司。
MK-7 标准品:质量分数为97.8%,批号为R080L0,美国默克公司。
脂肪酶:酶活不小于700 U/mg ,批号为BCCC7371,美国西格玛公司。甲醇、异丙醇:均为色谱纯,美国西格玛公司。无水碳酸钾:分析纯,中国国药集团化学试剂有限公司。
维生素K2保健食品样品:软胶囊、片剂,中国安琪纽特股份有限公司。
实验用水为符合GB/T 6682 规定的一级水。
1.2 实验方法
1.2.1 色谱条件
色谱柱:Intersil ODS-3 C18型柱(150 mm×4.6 mm,5 μm,日本岛津公司);流动相:甲醇-异丙醇(体积比为75∶25),流量为1.0 mL/min;柱温:40 ℃;进样体积:20 μL;检测波长:270 nm。
1.2.2 标准溶液的制备
MK-7 标准储备液:精密称取MK-7 标准品10 mg,置于100 mL 棕色容量瓶中,加异丙醇溶解并定容,摇匀即得质量浓度为100 μg/mL 的MK-7 标准储备液,于-18 ℃的冰箱中避光保存备用,临用前稀释至适宜浓度。
MK-7 系列标准工作溶液:分别吸取MK-7 标准储备液0.1、0.5、1、2、3、5 mL 于25 mL 棕色容量瓶中,加异丙醇定容至标线,摇匀即得质量浓度分别为0.4、2、4、8、12、20 mg/L 的MK-7 系列标准工作溶液,临用前配制。
1.2.3 样品溶液的制备
软胶囊样品溶液(含油脂):精密称取含油脂的软胶囊试样0.5~4 g(约相当于50~150 μg 的MK-7)于25 mL 棕色容量瓶,加入脂肪酶0.2 g、水10 mL,涡旋5 min,于37 ℃水浴4 h 以上。加入1 g 无水碳酸钾,涡旋2 min,冷却至室温,用异丙醇定容至25 mL,摇匀,以5 000 r/min 离心5 min,上层液体过0.22 μm 有机滤膜。
片剂样品溶液(不含油脂):精密称取不含油脂的片剂试样2 g(约相当于50~150 μg 的MK-7)于25 mL 棕色容量瓶,加入异丙醇15 mL,超声10 min,冷却,用异丙醇定容至标线,摇匀,过0.22 μm有机滤膜。
1.2.4 计算
MK-7 的质量分数按式(1)计算:

式中:X——样品中MK-7 的质量分数,μg/g;
β——由标准曲线查得试液中MK-7 的质量浓度,μg/mL;
V——定容体积,mL;
m——取样质量,g。
2 结果与讨论
2.1 样品前处理条件优化
以油脂为基质的MK-7 保健食品试样,易溶于酯溶性试剂异丙醇、正己烷等,参考木晓云等[3-5]的方法处理样品,即不经酶解直接提取后测定,发现样品溶液杂峰较多,干扰较大。主要因为辅料玉米油、大豆油或其它油脂在220~280 nm 波长范围内均有较强的紫外吸收,色谱分析时,辅料油会在MK-7 色谱峰前后产生较多的杂峰,无法分离。脂肪酶是将油脂水解成脂肪酸和甘油的酶,具有对油水界面的亲和力,并能在油水混合溶液中以高催化速率水解不溶于水的油脂。
本试验研究发现,当油脂量为4 g 时,加入0.2 g脂肪酶,以10 mL 水溶解,于37 ℃水浴4 h 以上可以将大部分油脂水解,加入碳酸钾可以与残留的少量油脂发生皂化反应,进一步减少油脂的干扰。异丙醇无毒,与水有较好的互溶性,保健食品中MK-7的添加水平较低,试样中MK-7 在异丙醇-水混合体系中可完全溶解,因此直接采用异丙醇定容,减少萃取、复溶等步骤,操作更简单。
不含油脂的试样则直接以异丙醇超声提取,使MK-7 完全溶解后进行色谱分析。操作简便、耗时较少。
2.2 专属性考察
制备阴性样品溶液进行HPLC 分析,考察空白溶液、阴性样品中的其它辅料对MK-7 测定的影响,其专属性考察结果见图1。标准溶液中MK-7 保留时间为14.043 min(图1a),软胶囊(含油脂)阴性样品溶液(图1b)、片剂(不含油脂)阴性样品溶液(图1d)均在14.043 min 处无色谱峰,说明辅料无影响,而含油脂样品溶液(图1c)、不含油脂样品溶液(图1e)在14.043 min 处出峰。MK-7 色谱峰理论塔板数为7 102,与其它杂峰分离度均大于2,证明方法专属性强。

图1 MK-7 色谱图
2.3 线性关系、检出限和定量限
分别吸取MK-7 系列标准工作溶液20 μL,进样,以质量浓度(x,mg/L)为横坐标,以色谱峰面积(y)为纵坐标,进行线性回归。结果表明,MK-7 的质量浓度为0.4~20 mg/L 的范围内与其色谱峰响应值线性关系良好,线性方程为y=26 801x+25 301,相关系数为0.999 8。取MK-7 对照品溶液适量,逐步稀释,用液相色谱仪记录其色谱峰高和信噪比,当信噪比为3∶1 时,MK-7 的检出限为0.06 mg/L。当信噪比为10∶1 时,MK-7 的定量限为0.2 mg/L。
2.4 加标回收与精密度试验
2.4.1 含油试样加标回收试验结果
准确称取不含MK-7 的维生素K2软胶囊阴性样品(内容物为除MK-7 的油脂试样)0.5 g 于25 mL 棕色容量瓶中,共9 份,加入脂肪酶0.2 g、水10 mL,涡旋5 min,于37 ℃水浴4 h。向样品中添加低、中、高3 水平MK-7 标准溶液,每水平平行测定3 次,加入1 g 无水碳酸钾,涡旋2 min,用异丙醇定容至标线,摇匀,以5 000 r/min 离心5 min,上层液体过0.22 μm 有机滤膜。按照1.2.1 色谱条件进行分析,以外标法计算溶液中MK-7 的浓度,并分别计算每份溶液的回收率,结果见表1。由表1 可知,含油试样的加标回收率为94.6%~97.9%,平均回收率为96.2%,测定结果的相对标准偏差为0.9%。
2.4.2 不含油试样回收试验结果
准确称取不含MK-7 的维生素K2片阴性样品1 g 于25 mL 棕色容量瓶中,共9 份,不经酶解,向样品中添加低、中、高3 水平MK-7 标准溶液,每水平3 次平行,加入15 mL 异丙醇,超声20 min,冷却定容至标线。摇匀,以5 000 r/min 离心5 min,上层液体过0.22 μm 有机滤膜。按照1.2.1 中的色谱条件进行分析,以外标法计算溶液中MK-7 的浓度,并分别计算每份溶液的回收率,结果见表1。
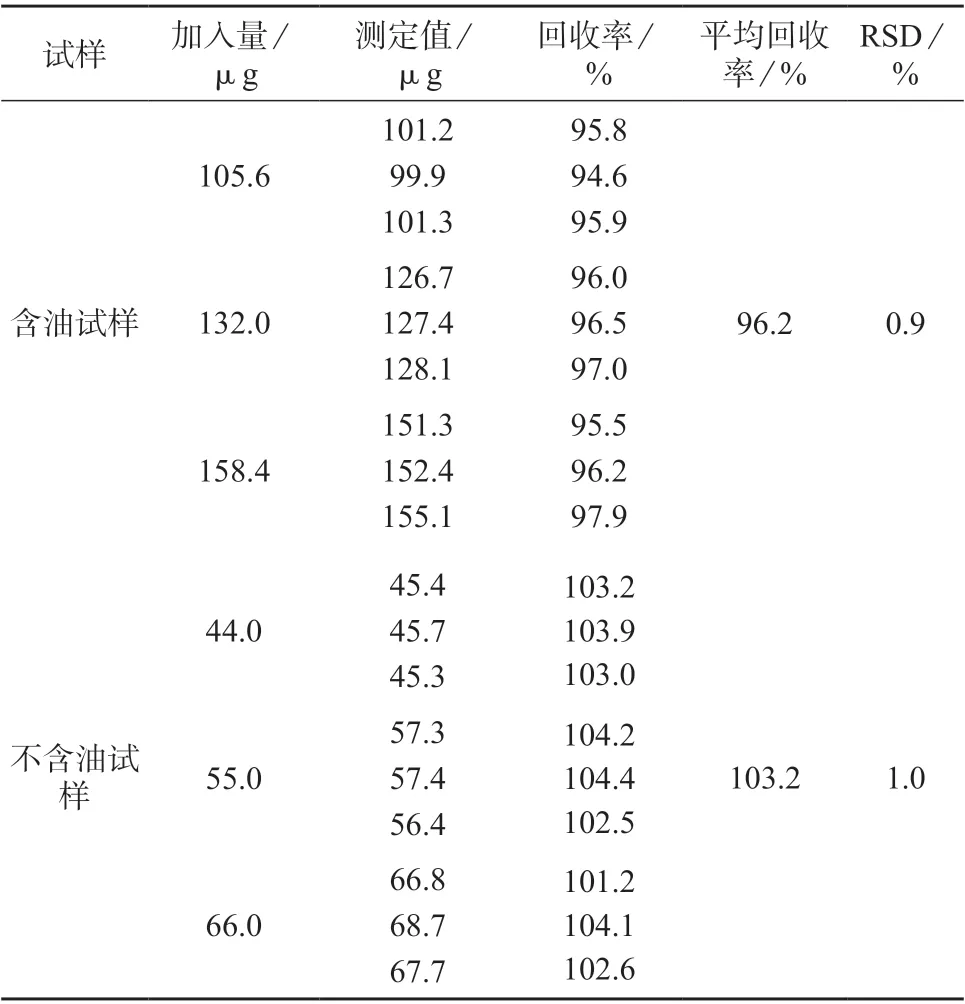
表1 MK-7 加标回收试验结果
由表1 可知,不含油试样的加标回收率为101.2%~104.4%,平均回收率为103.2%,测定结果的相对标准偏差为1.0%。
2.5 样品测定结果
取4 种含MK-7 的保健食品,分别按1.2.3 处理方法制备样品溶液,进行含量测定,测定结果列于表2。由表2 可知,实测值与投料理论值接近,说明本方法准确度较高。

表2 MK-7 样品测定结果
2.6 稳定性及耐用性试验
为考察样品溶液稳定性,按1.2.3 制备样品溶液,在室温条件下放置,分别在0、6、12 、24 h 测定其中MK-7 含量,考察含量是否发生变化。同时对色谱条件进行微小改变,如更换其它品牌C18色谱柱、柱温调整为35 ℃和45 ℃,测定含量考察方法耐用性,结果见表3。由表3 可知,样品溶液在24 h 内测定,以及色谱条件微小调整后测定,4 种样品的含量均未发生明显变化,表明方法耐用性良好。

表3 稳定性及耐用性试验结果
3 结语
曾尝试使用《GB 5009.158—2016 食品安全国家标准食品中维生素K1的测定》中的样品处理方法来测定维生素K2的含量,样品需要经过萃取、浓缩、复溶、衍生等步骤,而萃取过程易造成目标成分的损失,准确性低,样品处理往往需要多次萃取,还需要进行加标回收试验或使用内标的方法,以确定待测成分的损耗是否影响实验结果,操作复杂,耗时较长。
笔者经过试验,减少萃取等步骤,不含油脂的食品直接超声提取,含油脂食品通过酶解去除油脂的干扰后,以异丙醇直接提取,大幅缩短了检测耗时,提高了检测效率。测定结果的准确性经方法学考察,本方法专属性较强,具有良好的线性、精密度、准确度、稳定性。本方法不仅适用于MK-7 片剂保健食品,而且也适用于富含油脂的软胶囊类保健食品中MK-7 的含量测定。
猜你喜欢
中华胰腺病杂志(2022年4期)2022-08-23
天津医科大学学报(2021年4期)2021-08-21
求知导刊(2019年15期)2019-08-30
山东工业技术(2019年6期)2019-03-27
广西科技大学学报(2018年2期)2018-09-10
安徽农业科学(2018年4期)2018-02-02
科技创新导报(2017年20期)2017-09-13
建材发展导向(2016年6期)2017-01-17
科学与财富(2016年28期)2016-10-14
印刷技术·数字印艺(2015年10期)2015-12-10
