
热休克蛋白A12A改善内毒素血症肺损伤的实验研究
2021-09-07 09:38戴媛刘嘉莉闵新栩李蕴凡毛芊丁正年
实用老年医学 2021年8期
戴媛 刘嘉莉 闵新栩 李蕴凡 毛芊 丁正年
老年人感染发生率高,容易进展为脓毒症、感染性休克,导致多器官损伤和功能障碍,威胁病人生命[1]。肺脏是脓毒症/感染性休克的重要受累器官之一[2]。研究显示,感染导致的严重肺损伤死亡率高达27%,是ICU内死亡的独立相关危险因素[3]。因此,深入揭示脓毒症/感染性休克所致肺损伤的发病机制,对发现并鉴定新的有效治疗措施有积极意义。
热休克蛋白(heat shock proteins, HSPs)是一类进化保守的分子伴侣,根据分子量大小可分为HSPA/HSP70、HSPB/HSP27、HSPC/HSP90、HSPH/HSP110以及DNAJ/HSP40等几个亚家族[4]。一些HSP如HSP70、HSP90被证实可显著减轻脓毒症/感染性休克肺损伤[5-6],但尚未能成功临床转化,可能有更多其他HSP参与肺损伤的发生发展。
HSPA12A因其含有不连续ATP酶结构域,被认为是HSPA/HSP70家族的不典型成员。本团队的前期研究显示,HSPA12A在肥胖、非酒精性脂肪肝、肿瘤发生发展中具有重要作用[7-8]。但尚不清楚HSPA12A是否参与脓毒症、感染性休克肺损伤。本研究采用HSPA12A表达沉默的基因敲除鼠,探索HSPA12A在细菌脂多糖(lipopolysaccharide, LPS)所诱导的内毒素血症肺损伤中的作用。
1 材料和方法
1.1 实验动物 以C57BL/6为基因背景的Hspa12a基因敲除(Hspa12a-/-)鼠由本课题组前期构建完成[9]。本研究采用8~10周龄雄性小鼠用于实验,对照组采用一窝所生、同性别、野生型(WT)小鼠。实验动物饲养于南京大学模式动物研究所,并得到相应实验动物伦理学批准。
1.2 实验试剂 Escherichia coli LPS (0111:B4)、甲醛、抗甘油醛-3-磷酸脱氢酶(GAPDH)抗体为Bioworld Technology公司产品(St. Louis,MN,美国),抗HSPA12A抗体为Abcam公司产品(Cambridge,英国),High-sig ECL western blotting 底物为天能公司产品(上海,中国),BCA蛋白质测定试剂盒为Pierce产品 (Rockford,IL,美国)。
1.3 诱导内毒素血症 将WT鼠和Hspa12a-/-鼠分别分为2组,NS-WT组和LPS-WT组,NS-Hspa12a-/-组和LPS-Hspa12a-/-组(n=7)。LPS-WT组和LPS-Hspa12a-/-组进行LPS (5 mg/kg)单剂量腹腔注射,NS-WT组和NS-Hspa12a-/-组注射等体积生理盐水。6 h后进行相应分析。
1.4 血气分析 LPS或生理盐水腹腔注射后6 h, 以1.5%异氟烷麻醉小鼠,气管插管、机械通气,然后从左心室抽取动脉血液,用iSTAT Analyzer MN:300 (Abbott Park, IL,美国)进行血气分析。
1.5 组织学分析 LPS或生理盐水腹腔注射后6 h, 收集肺组织进行甲醛固定、石蜡切片以及HE染色,并在显微镜下观察(Olympus, 日本)。肺损伤评分按美国胸科协会制定的肺损伤量表进行评分[10]。见表1。
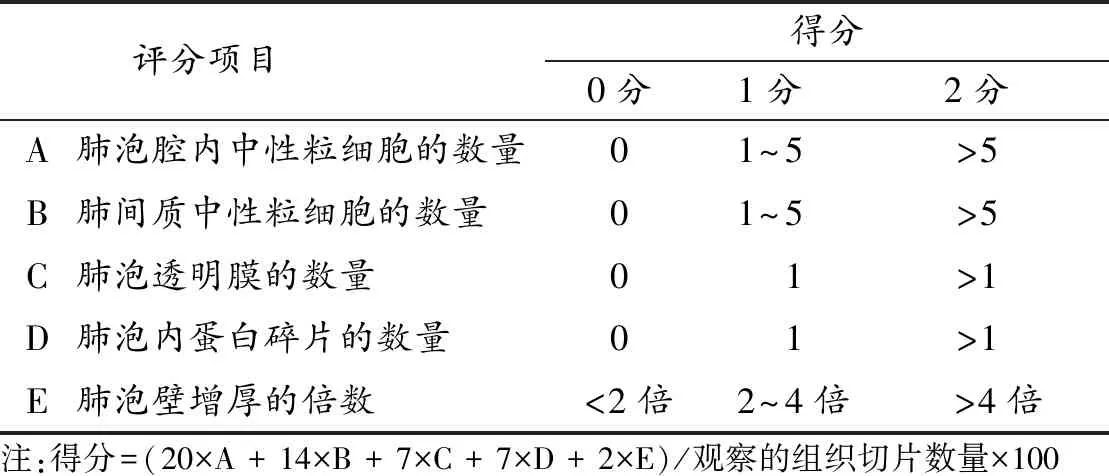
表1 肺损伤评分量表
1.6 免疫印迹(Western blot) LPS或生理盐水腹腔注射后6 h, 收集肺组织提取蛋白质,测定蛋白质浓度后,以等量蛋白质进行聚丙烯酰胺凝胶电泳,然后按顺序进行转印、封闭、孵育一抗、孵育二抗、显影。以抗GAPDH的印迹为上样内参。
2 结果
2.1 HSPA12A在内毒素血症小鼠肺组织中表达升高 本研究按照文献采用革兰氏阴性菌细胞壁主要致病成分LPS进行小鼠腹腔注射,诱导肺损伤的动物模型[11]。免疫印迹结果显示,LPS刺激使小鼠肺组织中的HSPA12A蛋白水平升高到对照组的1.5倍 (1.45±0.42比1.00±0.09,P<0.05)。见图1。

图1 NS-WT组和LPS-WT组HSPA12A蛋白水平比较
2.2Hspa12a基因敲除加重内毒素血症小鼠肺组织学改变 显微镜观察显示,WT鼠和Hspa12a-/-鼠的肺组织在LPS作用后均受到损伤,主要表现为肺泡腔和肺间质中的中性粒细胞浸润、肺泡内蛋白碎片沉积、肺泡壁增厚等病理改变。见图2。根据肺损伤量表进行评分后发现,LPS作用后,Hspa12a-/-鼠肺损伤较WT鼠显著加重 (P<0.01),见表2。
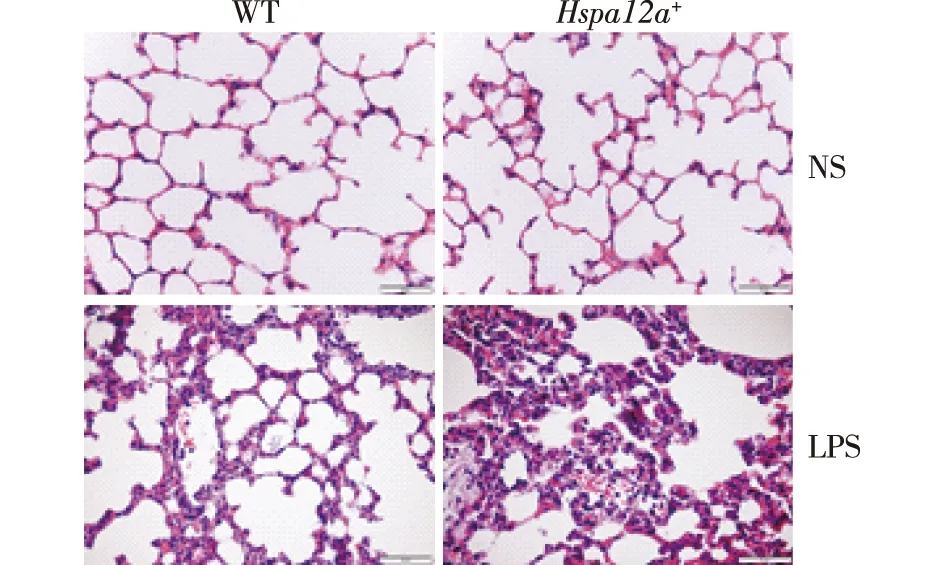
图2 WT鼠和Hspa12a-/-鼠的肺组织病理图像(HE,×20)
表2 小鼠肺组织肺损伤评分 分,n=6)
2.3Hpa12a基因敲除加重肺功能损害 与LPS-WT鼠相比,LPS-Hspa12a-/-鼠动脉血中总CO2(TCO2)以及CO2分压(PCO2)均显著升高 (P<0.05)。见表3。
表3 小鼠动脉血气分析
3 讨论
本研究发现,内毒素血症小鼠肺组织中HSPA12A表达水平升高,而Hspa12a基因敲除则加重内毒素血症肺损伤,提示HSPA12A在脓毒症/感染性休克肺损伤发生发展中具有重要作用。
感染所致肺损伤涉及多重信号网络形成的复杂病理生理过程,至今尚未完全阐明[12]。然而,肺泡壁水肿和肺泡内渗出造成的弥漫性肺损伤是其无可非议的、公认的关键病理特征[12]。此外,以中性粒细胞为主的白细胞聚集、纤维蛋白沉积也是此类肺损伤的常见病理改变。由于至少50%的脓毒症/感染性休克由革兰氏阴性菌引起,而LPS是位于革兰氏阴性菌细胞壁上的主要致病成分,也称内毒素,因此,LPS常被用于脓毒症/感染性休克的实验动物模型的诱导[13]。本研究给予小鼠5 mg/kg LPS单剂量腹腔注射,6 h后观察发现,小鼠肺组织出现肺泡壁间质和肺泡内白细胞渗出、肺泡壁水肿等典型的肺损伤病理改变,并且血气分析显示小鼠体内CO2过度蓄积,这些结果都提示小鼠出现了肺损伤。值得关注的是,上述LPS所致的肺组织损伤和肺功能损害在Hspa12a基因敲除鼠中显著加重,提示HSPA12A是保护脓毒症、感染性休克所致肺损伤所必需的。
现有研究证据显示,脓毒症/感染性休克肺损伤的病理改变主要与内皮细胞损伤、上皮细胞损伤、炎症细胞招募和浸润等因素有关。内皮细胞是肺泡壁的重要组成结构并形成血管屏障,控制分子、细胞进出,在肺泡气体交换、肺泡壁完整性方面的作用极其重要。LPS等细菌致病成分可导致内皮细胞凋亡(apoptosis)、焦亡(pyroptosis),从而损伤内皮屏障功能,导致肺泡蛋白成分、炎症细胞等渗出,而渗出的炎症细胞诱导炎症反应,产生大量炎症因子,对肺组织形成二次损伤。此外,肺上皮细胞损伤也是脓毒症/感染性休克肺损伤的重要病理改变。本研究结果显示,Hspa12a基因敲除可加重LPS所致的肺损伤,但其作用机制是否与内皮细胞、上皮细胞或炎症细胞有关有待进一步阐明。本团队前期研究显示,Hspa12a可调控癌变的肾小管上皮细胞增殖和迁移、肝脏巨噬细胞活化[7],提示HSPA12A在肺泡上皮细胞、炎症细胞的浸润和激活中可能也具有某种作用。此外,作为HSPA12A的高度同源基因HSPA12B,可显著抑制LPS所致的内皮细胞损伤、维持内皮屏障功能、限制内皮细胞与炎症细胞之间的交互作用[14]。结合HSPA12A参与脑卒中后血管新生的报道[9],提示我们未来值得从内皮细胞的角度探索HSPA12A对脓毒症/感染性休克肺损伤的保护机制。
综上,本研究发现Hspa12a是一个内毒素血症肺损伤的新型保护基因,但其作用机制尚有待进一步阐明。
猜你喜欢
体育科技文献通报(2022年4期)2022-10-21
中国药业(2022年12期)2022-06-29
中国典型病例大全(2022年12期)2022-05-13
昆明医科大学学报(2022年3期)2022-04-19
天津城建大学学报(2022年1期)2022-03-17
中国循证心血管医学杂志(2022年1期)2022-03-15
现代临床医学(2021年5期)2021-11-02
心电与循环(2021年4期)2021-08-03
医学前沿(2021年18期)2021-04-14
智富时代(2018年6期)2018-08-06
