
基于高原鼠兔简化基因组数据的微卫星引物开发
2021-08-03 06:37贺玉姣胡万军都玉蓉胡书立
野生动物学报 2021年3期
贺玉姣 胡万军 都玉蓉 王 婷 杜 雷 胡书立
(1.青海省青藏高原药用动植物资源重点实验室,青海师范大学生命科学学院,西宁,810008;2.青海师范大学体育学院,西宁,810008)
高原鼠兔(Ochotonacurzoniae)是一种小型哺乳动物(Mammalia),隶属于兔形目(Lagomorpha),鼠兔科(Ochotonidae),鼠兔属,主要生活在海拔3 000—5 100 m的高寒草原和高寒草甸,是青藏高原的优势种[1]。高原鼠兔一方面在维持青藏高原的生物多样性、草原生态系统中食物链的平衡等方面起到重要作用,被认为是青藏高原生态系统的关键种[2]。另一方面,在青藏高原的一些地区由于草地退化提高了啮齿动物(Rodentia)生境适合度,导致高原鼠兔种群密度过高,造成鼠害[3]。通过对高原鼠兔的种群遗传分析有助于了解该物种的种群结构,设立管理与保护单元,从而控制其种群数量[4]。而目前对高原鼠兔种群遗传的分析大多是通过线粒体基因进行的,基于核基因的相关分析则较少[5-6]。
SSR(simple sequence repeats)又称微卫星,是一类由几个核苷酸(1—6 bp)组成的串联重复DNA序列,广泛分布于真核生物和部分原核生物的核基因中[7]。SSR标记的高突变速率通常会产生高水平的多态性,同时它还具有进化所受选择压力小、共显性遗传、易于分析、较好的重复性等特点,使其广泛应用于遗传多样性、种群遗传结构等的研究中[8]。传统的SSR筛选方法,如富集文库筛选法,实验操作繁琐,筛选效率往往较低[9]。Li等[10]通过磁珠富集法对高原鼠兔的SSR进行分析仅得到13对引物。而随着高通量测序技术的发展,可以通过基因组、转录组等数据,对样本的SSR进行分析,得到大量SSR位点[11-13]。其中,简化基因组测序技术(RAD-seq)利用限制性内切酶对基因组 DNA 进行酶切,并对酶切片段进行高通量测序,该技术极大地降低了基因组的复杂度,不受参考基因组的限制,且测序成本低、准确率高[14],用此方法已经在大刺鳅(Mastacembelusarmatus)、印尼虎鱼(Datnioidespulcher)等动物中开发出SSR标记并应用于遗传研究[15-16]。本研究采用RAD-seq技术对高原鼠兔的SSR信息进行分析,并进行SSR引物的开发,为基于SSR分子标记开展的遗传多样性、种群遗传分析等研究提供基础资料。
1 材料与方法
1.1 样本与基因组DNA的提取
试验所用8个高原鼠兔样本采自青藏高原不同地区(表1),将鼠兔腿部肌肉组织置于2 mL离心管中,并用体积分数95%的乙醇保存。

表1 高原鼠兔采样位点信息
取约15 mg肌肉组织,用试剂盒(TIANamp Genomic DNA Kit,TIANGEN)提取基因组DNA,通过琼脂糖检测和NanoDrop 2000紫外分光光度计(Thermo Fisher Scientific,USA)对抽提DNA的质量和浓度进行检测。
1.2 简化基因组测序
取3个检验合格的基因组DNA样本送至上海欧易生物医学科技有限公司进行简化基因组测序,所用的测序技术为Super GBS,试验流程为:限制性酶切→连接接头→PCR扩增→混合建库→测序。
1.3 SSR位点信息分析与引物设计
对原始数据进行拼接、质控,然后用Stacks软件包(v 1.34)进行聚类,根据聚类结果进行组装过滤,得到最终的重叠群。利用MISA软件进行SSR位点搜索,搜索标准为单碱基、二碱基、三碱基、四碱基、五碱基、六碱基,重复次数分别在10、6、5、5、5、5次以上,相邻的SSR序列不少于100 bp。为了提高SSR的精度,取SSR与InDel的交集,条件为:①选取变异碱基数≥5的InDel;②InDel出现的位置位于SSR区域上下游5 bp内。然后利用软件Primer v3.2.5.0设计SSR引物。
1.4 引物验证
从设计的引物中随机挑选70对引物进行合成,合成时为了减少荧光引物合成成本,在正向引物的5′端添加1个M13接头序列。然后对8个高原鼠兔基因组DNA样品进行RCR扩增。PCR扩增体系为20.0 μL,包含2.0 μL 10×Taqbuffer(含Mg2+)(Takara,大连宝生物),5 U/μLTaqDNA聚合酶(Takara)0.2 μL,2.5 mmol/L dNTPs 2.0 μL,10 μmol/L上、下游引物各0.5 μL,0.2 μL总DNA,用灭菌超纯水补至20.0 μL。
反应参数设置为:95℃预变性8 min;95℃变性45 s,52—55℃退火45s,72℃延伸80s,35个循环;最后72℃延伸7 min。反应在伯乐热循环PCR仪S1000(Bio-Rad,Hercules,CA,USA)上进行。
对扩增结果进行毛细管电泳分型,利用GeneScan v3.1.2读取数据,并对数据进行整理、分析。
2 结果与分析
2.1 RAD-seq测序数据质量检测
高原鼠兔DNA的RAD-seq测序共获得原始数据2.14 Gb,对数据进行质控、过滤后得到高质量数据1.99 Gb(表2)。平均Q30(测序碱基质量值,即测序时错误识别的概率为0.1%、正确率为99.9% 的碱基比例)为89.11%,平均GC比例为50.47%,结果表明样本的数据量足够,测序数据的准确可靠性较高,GC比例正常。

表2 高原鼠兔DNA的RAD-seq基因组测序结果
2.2 简化基因组中SSR位点信息
由表3可知,通过MISA软件分析共检测到7 064个SSR位点,其中完全重复型(perfect)有6 806个,占位点总数的96.35%,而复合型较少,仅有258个,占总位点的3.65%。SSR位点中,单核苷酸重复基序最多,数量为3 260个,占总位点的46.15%;其次为二核苷酸重复基序,数量为2 710个,占总位点的38.37%;三核苷酸、四核苷酸和五核苷酸重复基序数量分别为535、256、40个,分别占总位点的7.57%、3.62%和0.57%;六核苷酸数量最少,仅有5个,占总位点的0.07%。在完全重复型SSR位点中,六核苷酸类型重复基序平均长度最长,可达到31 bp;单核苷酸类型重复基序的平均长度最短,为12 bp。在这些SSR位点中,单核苷酸重复SSR位点中A/T为优势基序,占该类型基序的98.22%;二核苷酸重复基序类型有11种,其中优势基序为AG/TC,数量分别为392、433个;三核苷酸重复基序有50种,AAC/TTG为主要基序;四核苷酸重复基序的种类数在完全重复型中最多,达83种,其中AAAC/TTTG为主要基序;五核苷酸和六核苷酸的重复基序类型数量分别为30和3,主要基序分别为AAAAC/CAAAA和CCTCAC/GCAGGA。
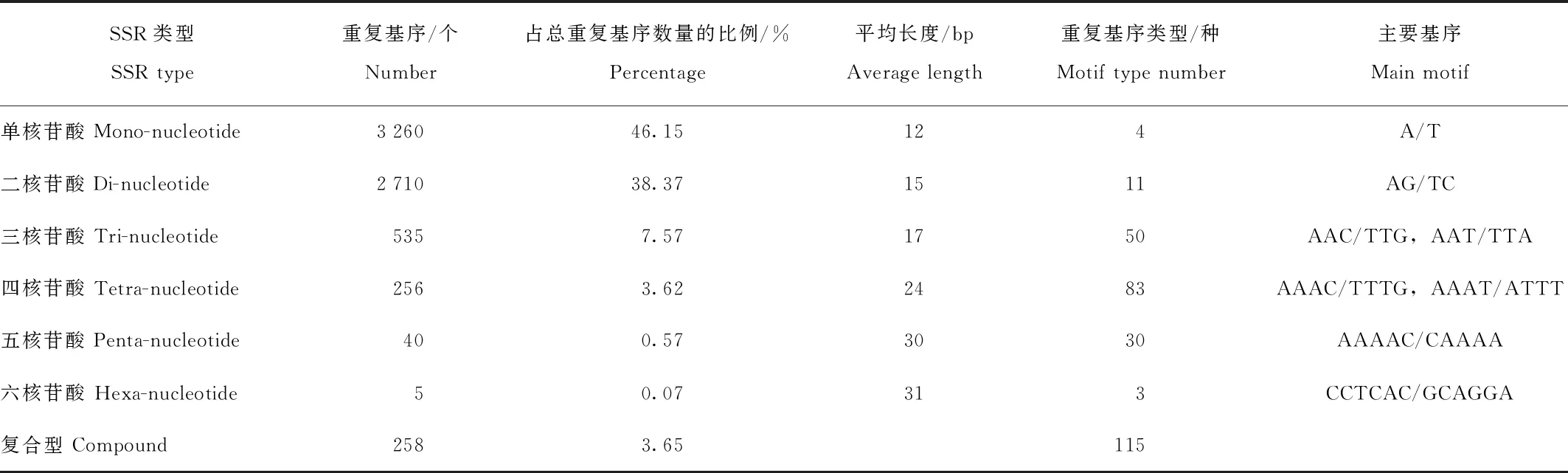
表3 高原鼠兔简化基因组中SSR位点信息
高原鼠兔基因组不同类型SSR位点基序的重复次数如表4所示。单核苷酸的基序重复次数主要集中在10—14次;二核苷酸基序的SSR主要集中在6—9次重复;三核苷酸基序的SSR主要为5—6次重复;四核苷酸、五核苷酸、六核苷酸基序的SSR均为4次重复。

表4 高原鼠兔不同类型及不同重复次数的SSR位点基序的分布数量
2.3 多态性SSR引物的筛选
在获得的7 064个高原鼠兔SSR位点中,通过取SSR与InDel的交集,获得182处符合条件的SSR区域,并最终设计出90对引物。随机选取70对引物,对8个高原鼠兔样本DNA进行扩增,其中无条带或主条带不明显的有7对,有条带但无多态性的引物有3对,有18对引物在8个DNA样本的检测中仅有2种多态性,其余42对引物具有单一条带,且多态性较好,占总检测引物的60%,引物具体信息如表5所示。

表5 42对具有单一条带的高原鼠兔多态性SSR引物信息

续表5
3 讨论
目前SSR标记的筛选方法主要有比较基因组法、微卫星富集文库法和基于组学的筛选法等,其中比较基因组法需要先获取近缘种微卫星位点信息,且不能筛选出该物种特有的微卫星,而富集文库筛选法前期需要进行SSR序列的富集及文库构建等复杂、繁琐的实验操作流程[17-18]。本研究使用简化基因组测序技术对高原鼠兔的SSR位点进行筛选,既简化了操作流程,同时位点的筛选效率也高于磁珠富集文库对高原鼠兔的筛选结果[10]。同为二代测序技术的转录组测序也被用于SSR标记的筛选中,通过转录组测序技术获得的SSR位点均位于基因的编码区,而SSR标记大多位于非编码区,且该区域的SSR标记通常具有更高的遗传变异,简化基因组序列可以均匀分布在物种的基因组中,通过该技术可以获得更多具有高变异SSR标记[19]。另外,无论与微卫星富集文库筛选法,还是转录组测序技术相比,RAD-seq所需费用都更低,因此该方法是针对目标物种进行特异性SSR标记筛选的经济、实用的方法。
在高原鼠兔的SSR位点中,数量最多的核苷酸重复基序是单核苷酸,其次是二核苷酸,这与Tóth等[20]在啮齿类动物中发现的二碱基重复基序数量最多并不一致。同时,高原鼠兔重复基序的类型也表现出一定的偏倚性,如二核苷酸重复基序中优势基序为AG/TC;三核苷酸中AAC/TTG为主要基序,这也与其他物种存在差异[16,21],而重复序列的数量与类型所表现出的差异可能都与物种特异性有关。Zane等[19]认为通常不将单碱基重复序列作为微卫星位点,应排除这一类型基序,但在本研究的验证实验中,有7对引物扩增的单碱基重复序列表现出明显的单一条带以及较好的多态性。
为节省荧光引物的设计成本,在验证实验中于正向引物上添加了M13接头,但该接头有可能会对引物的结构和结合特异性产生一定影响,导致扩增的失败[21]。同时由于验证样本仅有8个,使得剩余的18对引物的多态性还需要进一步验证,但总体来说用RAD-seq来筛选SSR标记时,在随机选取的70对引物中,成功获得了具有特异性单一条带,且多态性较好的引物有42对,占总数量的60%,表明具有较高的筛选效率。获得的42对引物足够满足高原鼠兔遗传多样性,种群遗传结构,谱系地理等问题的研究需求。
猜你喜欢
福建农林大学学报(自然科学版)(2022年4期)2022-11-01
世界科学技术-中医药现代化(2022年3期)2022-08-22
烟台大学学报(自然科学与工程版)(2022年3期)2022-06-30
肝博士(2022年3期)2022-06-30
南方医科大学学报(2022年3期)2022-04-13
水产科学(2022年2期)2022-03-20
南京师范大学学报(工程技术版)(2021年3期)2021-10-22
三农资讯半月报(2020年15期)2020-08-25
分子诊断与治疗杂志(2020年5期)2020-06-28
