
龙眼全基因组和转录本序列SSR位点的鉴定
2022-11-01 04:20林恩文林榕榕陈钦常徐秀明方静平
福建农林大学学报(自然科学版) 2022年4期
林恩文, 林榕榕, 陈钦常, 雷 雯, 徐秀明, 方静平
(1.福建师范大学生命科学学院,福建 福州 350117;2.厦门大学环境与生态学院滨海湿地生态系统教育部重点实验室,福建 厦门 361102)
龙眼(DimocarpuslonganLour.)起源于中国,为无患子科(Sapindaceae)龙眼属(Dimocarpus)乔木,常分布于亚热带地区,在我国已有2 000年的种植历史,主要种植于福建、广西等地区.得益于其独特的风味、丰富的营养和功能特效,龙眼广受人们喜爱,在我国具有一定的农业经济地位.我国具有丰富的龙眼种质资源(约400个品种[1]),占世界品种的2/3,开展龙眼群体遗传多样性以及种群遗传结构研究可以为龙眼资源遗传多样性保护提供理论基础.龙眼新品种(系)的培育主要是通过杂交育种,杂种鉴定保证双亲与后代亲缘一致是果树遗传改良的必要前提.传统的形态标记、细胞学标记和生化标记方法工作量大、周期长、经验性要求高、效率低下[2-3],已经不能满足品种快速鉴定的需求,而分子标记以其多态性强、信息量大、检测方便可靠[4],成为近年来杂种鉴定或品种遗传多样性分析最广泛采取的方法.
简单重复序列(simple sequence repeats, SSR)也称微卫星序列,广泛分布于真核生物基因组中,具有高可变性、显著多态性及相对的保守性、共显性等特点[5],已经在荔枝(Litchichinensis)[6]、红毛丹(NepheliumlappaceumL.)[7]、葡萄(VitisviniferaL.)[8]、菠萝[Ananascomosus(Linn.) Merr.][9]等果树的杂种真实性鉴定、种群遗传多态性分析、基因定位和遗传图谱构建等研究工作上取得成功.在龙眼SSR分子标记的研究中,陈虎等[10]利用SSR标记把龙眼划分为中国、泰国、越南等类群;洪自同[11]通过建立SSR引物和PCR扩增,鉴定了龙眼部分优良品种株系;胡文舜等[12]利用SSR鉴定了‘石硖’和‘香脆’两个龙眼品种正、反交的遗传多样性;洪仕南[13]鉴定了龙眼转录本SSR位点并探索了不同品种龙眼的亲缘关系.但以上工作均是基于龙眼基因组的部分信息,目前尚未有人展开对龙眼全基因组SSR位点的全面性挖掘和准确性鉴定.本实验室前期已完成了我国主栽早熟优质龙眼品种‘石硖’染色体级别高质量基因组的测序和组装工作(GenBank登录号:PRJNA741049),该工作组装了龙眼483.4 Mb的基因组序列,Scaffold N50为31.4 Mb,Contig N50为764 kb,覆盖龙眼全基因组99.30%的区域.基于该基因组序列,可以更准确全面地进行龙眼全基因组SSR序列鉴定和特征分析,从而筛选更准确的引物用于品种鉴定和遗传多样性分析.
本研究旨在挖掘具有优良基因型的‘石峡’龙眼品种的全基因组数据,结合生物信息学手段,鉴定龙眼全基因组的SSR位点,分析SSR位点在龙眼全基因组/转录本序列中的分布特征,包括基因组不同区域SSR位点的密度分布特征、不同长度基序的SSR分布规律等,并对单、双子叶植物及无患子目等不同种的植物进行全基因组水平的SSR位点鉴定和比较,总结SSR位点的一般规律和物种特异性.本研究旨在为龙眼的真实杂交种鉴定、遗传多样性研究、遗传图谱构建和分子标记辅助育种提供重要数据库支撑,对其他物种SSR位点的深度挖掘和鉴定也提供参考和方向.
1 材料与方法
1.1 RNA提取、RNA文库构建与测序
选取龙眼不同时期的花芽、叶芽、果实进行转录组测序,每个处理设计3个生物学重复.采用天根RNA提取试剂盒[Qiagen, #74904,天根生化科技(北京)有限公司]提取龙眼不同样本的RNA,提取方法见试剂盒说明书.采用DNA纯化试剂盒 TIANquick Midi Purification Kit[天根生化科技(北京)有限公司]去除RNA中的DNA污染.RNA样品建库后在Hiseq×10 测序仪(Illumina公司)上进行双端测序,测序模式为“快速模式2×150 nt”.
1.2 基因组数据和转录组数据
龙眼品种‘石硖’染色体水平的基因组已在NCBI公布,GenBank登录号为:PRJNA741049.该基因组数据用于下游SSR的挖掘.通过TRINITY软件(http://trinityrnaseq.github.io/)对前期获得的所有转录组数据进行拼接,得到转录本序列,该数据总大小约为48.6 Mb.
1.3 SSR的挖掘
利用Perl软件编写的一款软件MISA扫描和挖掘‘石硖’龙眼全基因组的SSR序列,这款软件的安装包下载于http://pgrc.ipk-gatersleben.de/misa/.在配置文件参数的设定中,考虑了完美和复合标记类型.核苷酸重复基序长度设置为2~6 bp,二、三、四核苷酸SSR序列的最短长度均为12 bp,五、六核苷酸SSR序列的最短长度分别为15、18 bp.在本研究中,单核苷酸的SSR位点未被考虑,因为基因组序列的测序错误和组装错误常常会引入新的单元重复,多聚腺苷酸尾巴 (polyA)也常与单核苷酸重复混淆.对于复合SSR重复序列,两个SSR位点之间的最大差异设为100 bp.使用在线gff2bed Python脚本(http://bedops.readthedocs.org/en/latest/content/reference/filemanagement/conversion/gff2bed.html)、bedtools软件(https://bedtools.readthedocs.io/en/latest/index.html)的“bedtools getfasta”命令和自编的Python脚本将3′UTR区、5′UTR区、CDS区、外显子区和内含子区从gff3注释文件提取出来.利用Windows Excel 2010软件和自编的Python脚本对龙眼全基因组、基因区和EST序列的SSR密度、G+C含量及核苷酸重复基序、重复长度、重复次数分布进行了估计和分析,并进行了比较.互补链上的重复基序被认为是等价物,并组合成一个基序,如AG基序相当于GA、TC、CT等.
1.4 龙眼SSR相关基因的鉴定
为了鉴定SSR位点相关基因的功能,通过编写的Python脚本提取包含SSR位点的基因.利用在线网站(http://eggnog-mapper.embl.de/)对龙眼全基因组做基因本体联合会(gene ontology,GO)注释,将含有SSR位点的基因作为目的基因,全基因组基因作为背景基因,进行GO富集分析.
1.5 近缘物种系统发育树的构建
近缘物种系统发育树的构建一般基于单拷贝基因,利用OrthoFinder软件(https://github.com/davidemms/OrthoFinder)鉴定了不同物种间的单拷贝基因,得到了龙眼与其近缘种荔枝、红毛丹、文冠果(XanthocerassorbifoliumBunge)、漾濞枫(Aceryangbiense)的单拷贝同源基因列表信息,通过EasySpeciesTree脚本(https://github.com/dongwei1220/EasySpeciesTree)构建系统发育树.
2 结果与分析
2.1 SSR在基因组不同区域中的密度分布
基于最新组装的483.4 Mb龙眼基因组,共挖掘到260 204个SSR位点,SSR平均密度为每1 Mb含有538.24个SSR位点(即每1 kb含有1.86个SSR位点,不包含单核苷酸SSR).其中,39 102个(15.03%)分类为类型一SSR(≥20 bp),密度为每1 Mb含有80.88个SSR位点.221 102个(84.97%)分类为类型二SSR(≥12 bp,且<20 bp),密度为每1 Mb含有457.35个SSR位点.类型二SSR的数量和密度远超类型一SSR.基因区(含外显子区、内含子区、CDS区、5′UTR区、3′UTR区)和转录本中SSR数量和密度的分布如附图1(扫OSID码可见)、表1所示.非编码区SSR的密度显著高于编码区.在非编码区中,5′UTR区SSR的密度最高,每1 Mb含有955.47个SSR位点,接着依次为基因间区(每1 Mb含有567.39个SSR位点, 见附图1, 扫OSID码可见)、内含子区(每1 Mb含有535.15个SSR位点)、3′UTR区(每1 Mb含有385.88个SSR位点).CDS区SSR的密度最低,每1 Mb仅含有240.36个SSR位点.
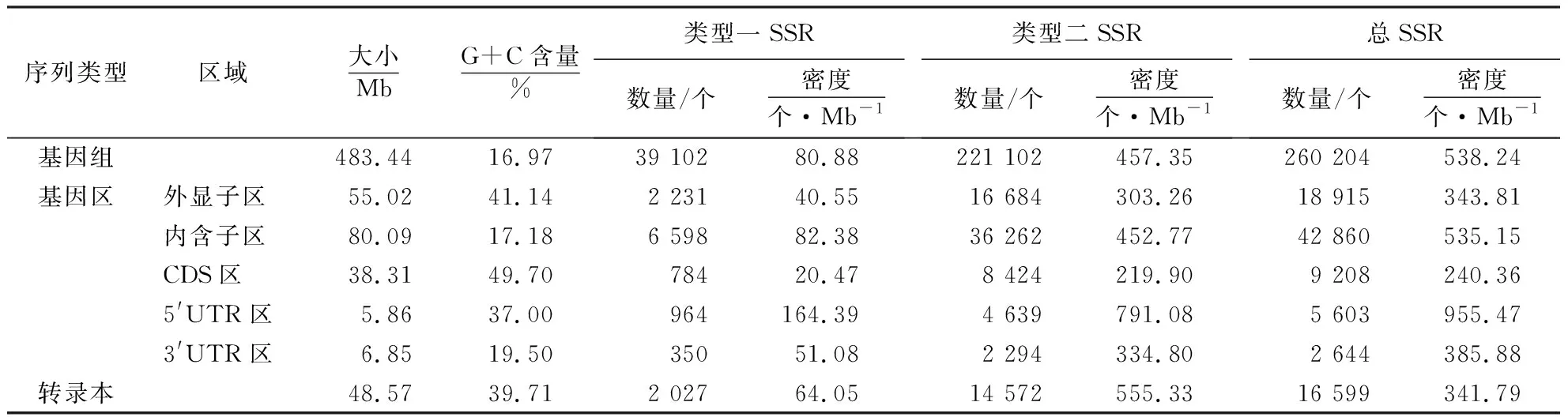
表1 龙眼不同基因组区域和转录本序列中SSR密度、G+C含量的分布Table 1 SSR density and G+C content in different genomic regions and transcript sequences of longan
2.2 SSR总长度与基序长度的分布
在全基因组中,SSR重复序列总长分布为12~69 bp,而转录本SSR重复序列总长分布为12~309 bp.基因组和转录本的SSR重复序列长度均以12 bp为主,分别占SSR总量的48.26%、52.00%(图1a),分别占类型二SSR总量的56.80%、59.24%(图1b).排在第2位的SSR重复序列长度为15 bp,其占比在基因组和转录本中的趋势是相似的,在基因组中约13.33%的SSR长度为15 bp,在转录本中,有15.27%的SSR长度为15 bp.在全基因组的类型一SSR中, 总长为20 bp的重复序列最多,为11 533个(29.49%),其次是24 bp(17.70%)(图1c).相反地,在转录本中24 bp的SSR重复序列最多,为497个(24.52%),紧接着是20 bp(22.00%).有趣的是,当SSR重复序列长度为3的倍数时(如12、15、18、21、24、27、30 bp等),转录本对应的SSR数量高于基因组中的SSR数量.
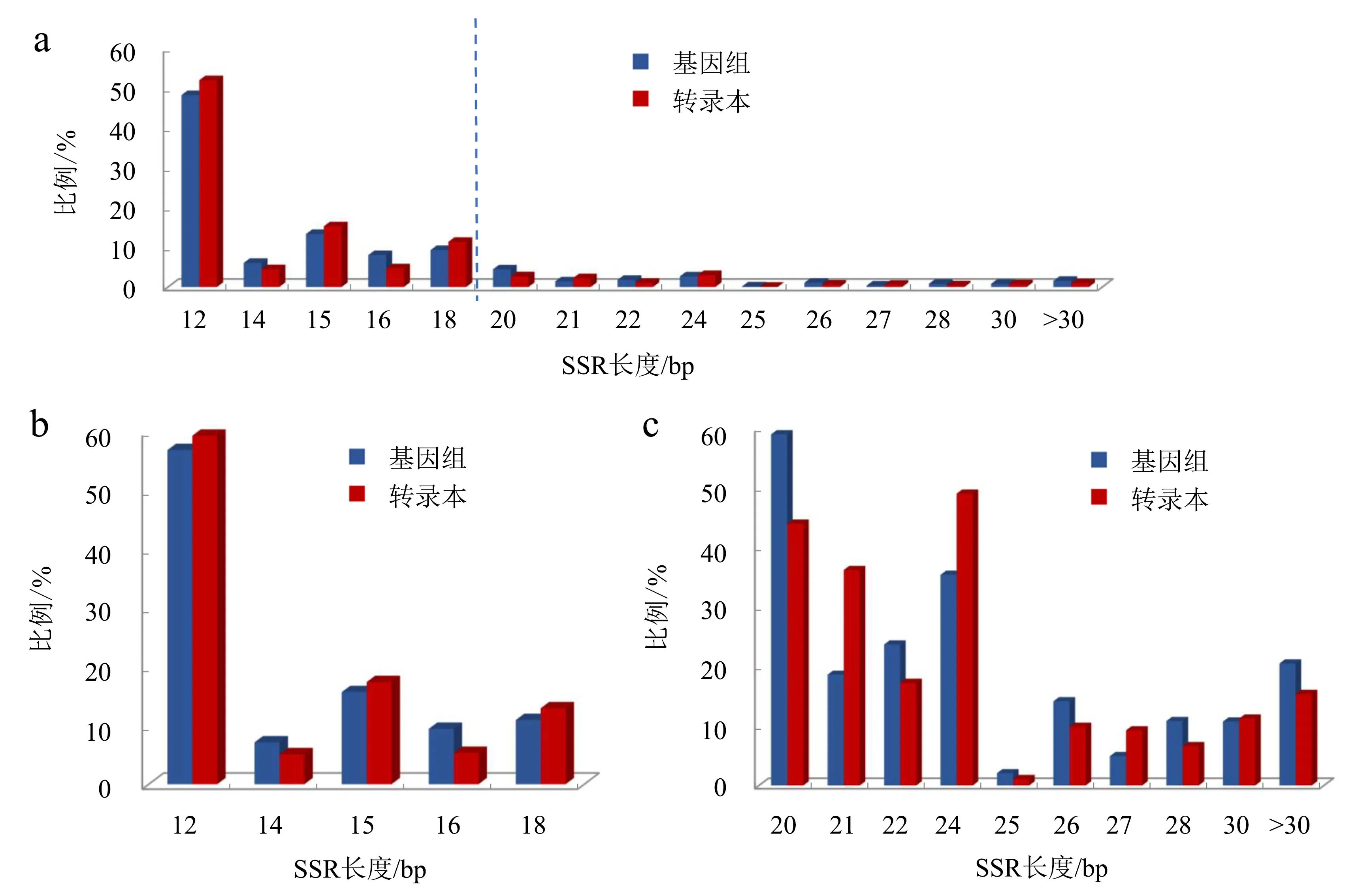
a:全基因组;b:类型二SSR;c:类型一SSR.图1 SSR长度在龙眼全基因组和转录本序列中的分布Fig.1 Distribution of longan SSR at the genome and transcriptome level by repeat length
不同SSR基序长度的分布如图2所示.在全基因组中,二核苷酸基序SSR的数量最多,为86 048个(33.07%),大于其他基序(图2a).类型一中的SSR以二核苷酸基序为主(图2b),高达60.95%,数量23 833个,接着依次为三、四、五核苷酸基序,分别占SSR总量的19.58%、7.58%、8.61%.在基因组和类型一的SSR中,SSR数量随核苷酸基序长度的增加而减少.但在类型二SSR中却没有呈现该规律(图2c),类型二SSR中重复次数最多的为四核苷酸基序,为65 853个(占29.78%),略高于三核苷酸基序和二核苷酸基序(分别占28.57%、28.14%).在全基因组中,SSR不同基序分布密度在CDS区、外显子区、内含子区和UTR区呈现不同的趋势(图2).基因编码区的分布密度与非编码区显著不同,编码区(含转录本、CDS区、外显子区)的三核苷酸基序数量明显高于其他核苷酸基序,且六核苷酸基序高于五核苷酸基序.转录本、CDS区和外显子区的三核苷酸基序数量占比分别为52.51%、79.19%、56.51%,是非编码区5′UTR区、3′UTR区和内含子区的该基序占比的1.84倍以上(分别为28.52%、28.29%、22.89%)(图2a).在转录本SSR中,三、六核苷酸基序比例略微高于基因组中的该基序(图2).
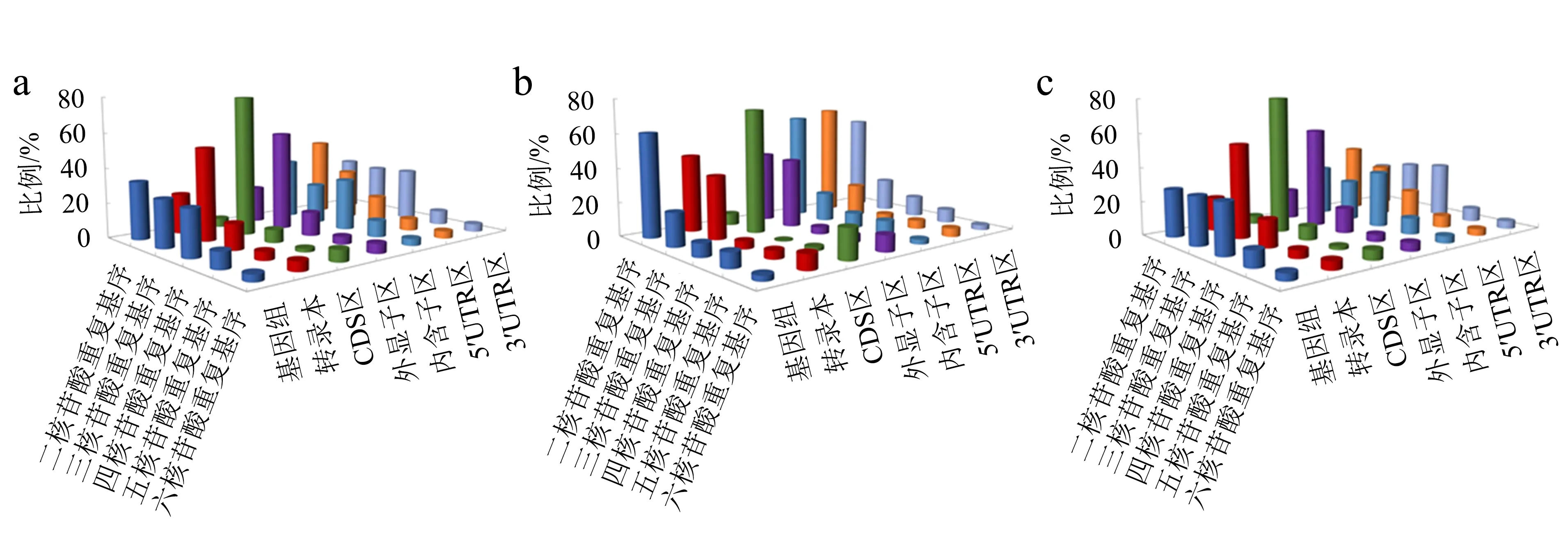
a:全基因组;b:类型一SSR;c:类型二SSR.图2 不同SSR基序长度在龙眼全基因组和转录本序列中的分布Fig.2 Distribution of longan SSR at the genome and transcriptome level by motif length
2.3 SSR基序在全基因组和转录本中的分布
在全基因组和转录本中,不同基序的重复次数频率随重复单元数量的增加而急剧下降(图3).在全基因组和转录本中,以二核苷酸基序为重复单元的SSR位点的平均数量(每1 Mb含有177.99个SSR位点)为三核苷酸重复基序的1.74~1.76倍,是四至六核苷酸重复基序的2.53~2.73倍(附表1,扫OSID码可见).在基因组区域中,二核苷酸重复基序的累计长度为1 472.40 kb,是所有核苷酸重复基序类型中最长的.在转录本中,尽管二核苷酸重复基序SSR平均重复单元数量(8.23个)是三核苷酸重复基序SSR(4.74个)的1.74倍,然而由于三核苷酸重复基序的出现频率(8 716次)最高(52.51%),分布密度最高(每1 Mb含有179.47个SSR位点),因而三核苷酸重复基序在转录本SSR中所占比重最大:其累计长度为123.89 kb,约是二核苷酸重复基序SSR累计总长(62.33 kb)的1.99倍(附表1,扫OSID码可见).在龙眼基因组中最长的SSR是以AAG三核苷酸为重复基序的序列,重复次数为103,长达309 bp;其次仍是以AAG三核苷酸为重复基序的序列,重复次数为99,长达296 bp.在转录本中,二核苷酸重复基序序列最长,以CT二核苷酸为重复基序的序列,重复次数为34,累计长度为68 bp;其次是以AGA三核苷酸为重复基序的序列,重复次数为18,长度为54 bp.在基因组和转录本相同的重复基序中,重复基序的出现次数和序列长度均不同,更详细的信息见图3、附表2(扫OSID码可见)、附表3(扫OSID码可见).
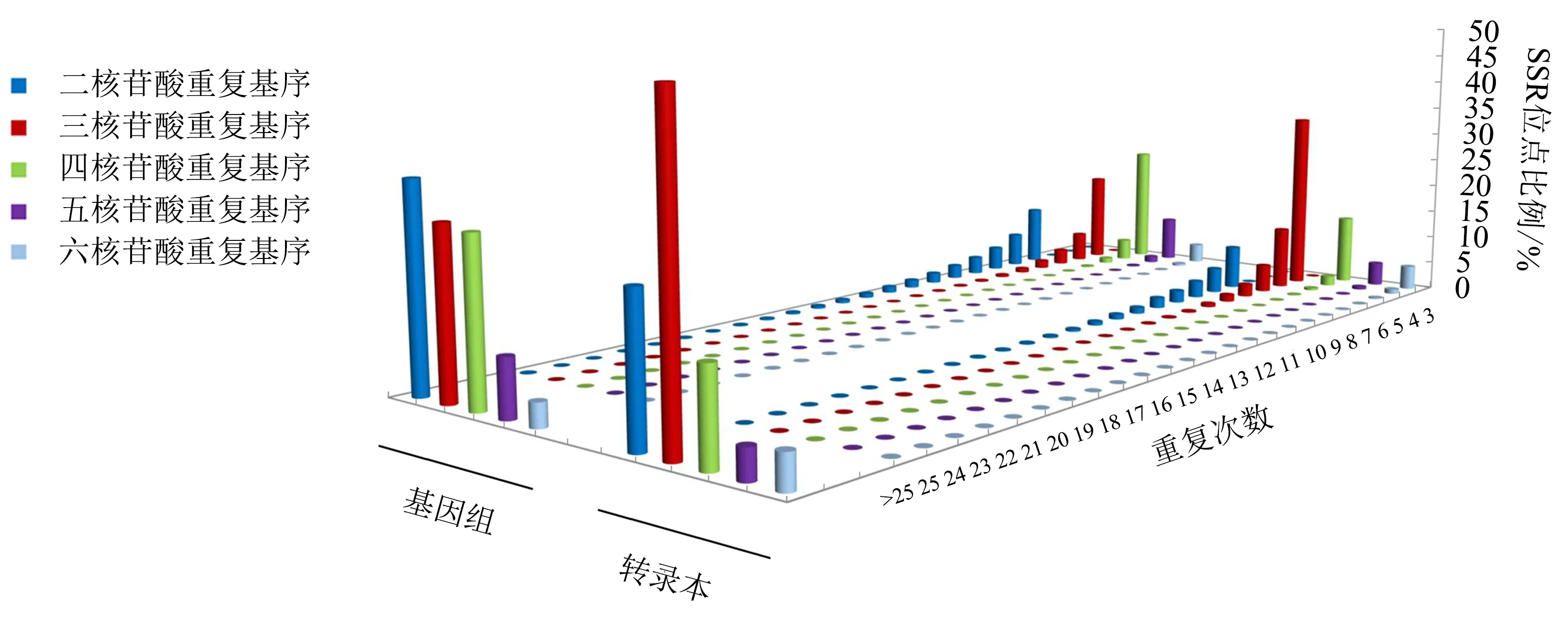
图3 龙眼全基因组和转录本序列中SSR重复单元和基序的分布Fig.3 Frequency of longan SSR at the genome and transcriptome level by motif length
2.4 含有SSR重复基序的基因鉴定
本研究结果表明:在含有SSR位点的基因中,含有1~10个SSR位点的基因数量最多(附图2a,扫OSID码可见);在这些基因中,SSR位点分布在外显子上的数量集中在1~4个(附图2b,扫OSID码可见).共有20 761个基因含有SSR位点,占全基因组基因数量的56%,不含SSR位点的基因数量为16 381个(44%)(附图2c,扫OSID码可见).不同基因含有不同数量的SSR位点.含有SSR位点的基因中,一个基因最多含有42个SSR位点,8 761个基因(42.19%)只含有一个SSR位点,5 011个基因(24.14%)含有两个SSR位点,2 813个基因(13.55%)含有3个SSR位点.在不同的基因组区域中,内含子片段包含的SSR位点最多(24 981个),外显子片段包含的SSR位点最少(1 730个)(附图2d,扫OSID码可见),含有SSR位点的外显子数量随SSR位点的增多而急剧下降(附图2b,扫OSID码可见).
将含有SSR位点的基因进行GO注释富集分析,结果如附图3(扫OSID码可见)所示.在20 761个含有SSR位点的基因中,9 231个基因可比对到一个或多个GO号,分别涉及到1 129个生物过程、2 442个细胞组分和7 476个分子功能.在分子功能分类中,较高比例的基因(66.70 %)被富集到蛋白结合和催化活性(62.00%)等功能,接着是转运活性(25.36%)和核酸结合转录因子活性(10.23%);在细胞组分分类中,97.21%的基因被注释到细胞,接着是细胞膜/细胞器或部分细胞器和分子复合物,所占比例分别是40.26%、72.85%(36.74%)、17.38%;在生物学过程分类中的基因占比前三的数据如下:注释到细胞类别中的比例为85.73%,单组织过程中的比例为71.06%,代谢过程中的比例为62.66%;其他主要被注释到的功能是:生物调节、应激反应及细胞成分与信号传导等.
2.5 龙眼与近缘种SSR位点数量和分布的比较
基于NCBI下载的无患子目无患子科的3个高质量基因组(荔枝、红毛丹、文冠果)和无患子目槭树科的漾濞枫等物种已组装的高质量基因组,使用与本研究中龙眼SSR位点鉴定相同的方法对其做了全基因组SSR位点的鉴定,并分析了不同基序SSR位点的分布规律,结果如表2、附表4(扫OSID码可见)、图4所示.龙眼与荔枝基因组大小仅相差13.04 Mb,SSR数量分布特征和G+C含量特征最为相似,其次相似的是红毛丹.通过比较5个物种SSR的分布,漾濞枫基因组最大(666 Mb),所含SSR位点的数量最多(457 104个);文冠果基因组大小位居第二(506 Mb),但文冠果是5种植物中含有SSR位点数量最少的物种(附表4,扫OSID码可见).基于拷贝同源基因构建的进化树,龙眼、荔枝与红毛丹有着较近的亲缘关系,龙眼与荔枝的亲缘关系最近,而与文冠果、漾濞枫的亲缘关系较远(图4a).通过统计5种植物不同基序SSR位点的重复次数,结果(图4b)显示,二、三核苷酸重复基序的重复次数均高于其他基序类型.通过统计5种植物不同基序长度SSR位点的G+C含量,结果(表2)显示,除漾濞枫外,二至六核苷酸重复基序的G+C含量(12.43%~25.17%)远低于A+T含量(87.57%~74.83%).在漾濞枫的六核苷酸重复基序中,G+C含量显著上升为36.01%,其他植物六核苷酸重复基序的G+C含量为18.63%~24.04%.
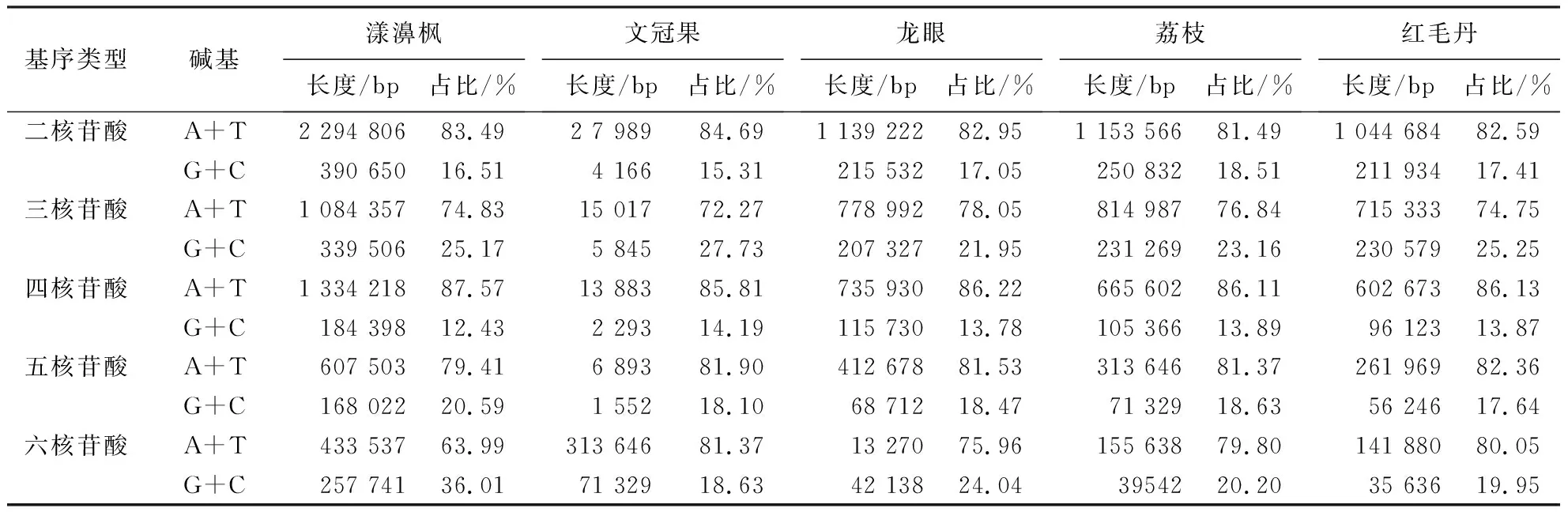
表2 不同物种不同基序G+C含量的统计Table 2 G+C content of different motifs in longan and closely related species
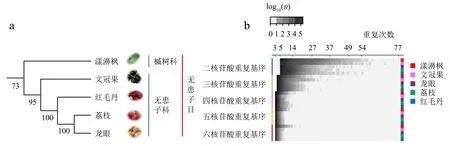
a:系统发生树;b:SSR分布数量热图.图4 龙眼近缘物种系统发生树和SSR分布数量热图Fig.4 Phylogenetic tree and heatmap of SSR in longan and closely related species
2.6 单、双子叶植物SSR位点的比较
本研究还比较了不同单、双子叶植物中SSR位点的数量分布规律和G+C含量分布情况.除了双子叶植物龙眼外,还选取拟南芥(Arabidopsisthaliana)、橙子(Citrussinensis)、葡萄3种双子叶植物及水稻(OryzasativaL.)、高粱[Sorghumbicolor(L.) Moench]两种单子叶植物.分别对这些物种的基因组和转录本进行SSR位点鉴定和比较分析,结果如附表3(扫OSID码可见)所示.单子叶植物基因组SSR比双子叶植物呈现出较高的G+C含量.在单子叶植物的基因组中,水稻和高粱SSR的G+C含量分别为43.60%、43.90%;而在双子叶植物中,龙眼SSR的G+C含量仅为16.97%,拟南芥、橙子和葡萄SSR的G+C含量分别为36.00%、32.30%、34.40%.单子叶植物水稻和高粱的SSR均是以三碱基重复单元为优势重复单元,水稻SSR的三碱基重复单元分布密度最大,每1 Mb含有220.1个SSR位点;在双子叶植物中,除拟南芥以三碱基重复单元为优势重复单元外,其他物种的SSR均是以二碱基重复单元为优势重复单元,橙子的二碱基重复单元密度最大,每1 Mb含有146.0个SSR位点.在转录本SSR中,单子叶植物依旧具有较高的G+C含量,水稻和高粱的G+C含量分别高达51.50%、52.00%;而在龙眼、拟南芥、橙子和葡萄等双子叶植物中,G+C含量均表现出明显的下降趋势,分别为39.71%、42.70%、41.40%、43.90%.与基因组SSR不同,在转录本SSR中,本研究涉及的所有单、双子叶植物均是以三碱基重复单元为优势重复单元,但是在单子叶植物中,四至六碱基重复单元的密度比双子叶植物约高出一倍,即单子叶植物的SSR密度均大于双子叶植物.
3 讨论
微卫星标记在遗传学、生态学、分类学和进化研究中起着至关重要的作用.分析龙眼编码区和非编码区的SSR位点,有助于理解龙眼基因组结构与进化,为揭示微卫星标记在基因调控中的潜在作用以及基因定位提供数据支持.随着近期龙眼基因组的公布,可对龙眼SSR位点进行全基因组水平鉴定.本研究首次对龙眼高质量基因组进行SSR位点分析并总结其分布规律,为开展龙眼属植物的杂种鉴定、遗传图谱构建、种质资源保护等提供了重要的基础数据库.本研究还挖掘了SSR位点在龙眼转录本中的分布情况,首次对龙眼SSR序列全基因组和转录本内部特征分布进行了详细的比较分析.
在龙眼基因组中,以二核苷酸重复基序为优势基序,SSR位点的分布频率与重复单元数量呈反比,这符合一般规律[14],与菠萝[9]、石榴(PunicagranatumL.)[15]、闽楠(Phoebebournei)[16]等物种的分析结果一致.但在禾本科植物中没有此规律,它们以三、六核苷酸为优势重复类型[17].本研究也发现,单子叶植物(水稻、高粱)基因组中常常以三核苷酸重复为优势,双子叶植物(龙眼、橙子、葡萄)往往以二核苷酸重复为优势.已有研究显示,双子叶植物的SSR位点在基因组出现的频率要比单子叶植物高得多[18],但本研究未发现相同的趋势.
在龙眼基因组中,类型二SSR的数量比类型一SSR更丰富(图1),该规律与其他物种[19-20]类似.本研究中,龙眼基因组共鉴定到了260 204个SSR位点,SSR平均密度为538.24个·Mb-1,漾濞枫、红毛丹、文冠果和荔枝SSR的平均密度分别为686.34、678.01、415.92、545.09个·Mb-1,荔枝与龙眼的亲缘关系最近,基因组大小及SSR位点的数量和分布最相似.本研究还将无患子目目前已有的参考基因组构建了进化发生树,尽管红毛丹的外形、口感与荔枝相似,但该进化树表明了无患子科的荔枝与龙眼的关系近于红毛丹,这与前人利用同源序列分析研究的结果[21]一致.另外,文冠果与龙眼的亲缘关系最远.据已有研究报道,二核苷酸重复或三核苷酸重复单元越多,则表明该物种具有较高的进化水平,反之,则物种的变异较少或进化时间短[22].漾濞枫基因组合中含有最多的二、三核苷酸重复基序,暗示了漾濞枫在无患子目中可能具有较高的进化水平.甘蔗(Saccharumofficinarum)、玉米(ZeamaysL.)、高粱等禾本科植物SSR数量的多少与基因组大小呈正相关[23],而无患子科却没有此规律.
基因不同区域SSR密度的分布规律与以往的研究报道[20]基本一致.不同基因位置的SSR可能在发育、适应、生存和进化中发挥不同的作用.含有SSR位点的基因区域的突变可能会影响相应的基因产物.如:基因编码区SSR位点的插入或缺失可能通过移码突变或扩展毒性mRNA导致基因功能的获得或丧失[24];在UTR或内含子中存在某些多态SSR可能会影响基因的表达水平[25].通过比较龙眼转录区域(CDS、外显子、EST)和整个龙眼基因组区域的SSR位点的结果表明,除三、六核苷酸重复外,所有重复类型在转录区域的数量都相对较少[图2、附表1(扫OSID码可见)].这种趋势在其他物种中也存在.龙眼转录本中以三核苷酸重复基序为优势基序,三、六核苷酸相对于其他重复类型的优势被归因于对移码突变的负选择.三、六核苷酸是多个密码子的整合,它们的突变可能不会破坏阅读框,这一过程可能与遗传保护有关[20].以往的研究显示,在睡莲(Nymphaeatetragona)[26]、荔枝[27]、黑莓(RubusfruticosusPollich)[28]、胡萝卜(Daucuscarotavar.sativaHoffm.)[29]的转录本中,二核苷酸重复基序的SSR数量最多,在柑橘(CitrusreticulataBlanco)[30]、党参[Codonopsispilosula(Franch.) Nannf.][31]、芒果(MangiferaindicaL.)[32]中是三核苷酸重复基序的SSR数量最多,因此在转录本中依旧是以二、三核苷酸重复基序为优势基序.在龙眼中,全基因组序列SSR以二核苷酸重复基序为优势基序,转录本SSR以三核苷酸重复基序为优势基序,基本具有基序越短而重复次数越多、基序越长而重复次数越少的规律.此外,对龙眼转录本SSR的挖掘,有利于进一步进行重要功能基因的关联,做更精细的基因定位.徐志军等[33]通过转录本数据和SSR位点图谱,联合重要性状的QTL位点对抗病基因做精准定位,这是今后研究可探讨和结合的方向.
SSR位点区域被认为是基因组中的突变热点区域,因此在基因组的起源和进化动力学中发挥着重要作用[34].这一趋势可能是由于较长的类型一SSR重复序列固有的不稳定性,这些重复序列容易通过复制滑移、点突变或重组等机制突变为不完美的SSR[34],而较短的类型二SSR重复序列对突变具有更强的耐受力而被保留.在本研究所有物种的SSR位点中,均是AT占主要优势,而G+C含量较少,这种现象在动植物中普遍存在.如在花生(ArachishypogaeaLinn.)的SSR位点中,主要重复单元也是AT、GA、GT和ATT[35].若假设SSR区域是DNA稳定区域,GC富集区域也相对稳定,只存在较少的突变,那么AC或AG的重复应多于AT,但本研究却发现相反的情况,这暗示着AT与GC富集基序的突变或修复机制的差异,如打破AT结构类型的碱基所需的能量相对于GC结构类型的碱基需要的能量更少,使得AT更容易产生突变[36].本研究也观察到,随着核苷酸重复基序变长,AT优势呈下降趋势.
SSR在植物基因中的功能尚不清楚.本研究对龙眼含有SSR位点的基因进行功能注释和分类的结果表明,这些基因具有一系列功能,如蛋白质结合、催化活性、代谢酶、疾病信号转导、结构和存储蛋白、转录因子等.在分子功能分类中,含有SSR位点的基因大多与具有结合和催化活性的蛋白质同源;在细胞成分分类中,多与细胞、细胞膜和细胞器相关.表明SSR在植物代谢、基因表达调控和基因进化中可能具有重要的生物学意义.
猜你喜欢
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
电脑报(2020年40期)2020-11-06
医学信息(2019年4期)2019-10-08
作文大王·低年级(2019年4期)2019-05-13
烹调知识(2019年3期)2019-03-01
世界热带农业信息(2018年5期)2018-11-09
电脑知识与技术(2018年19期)2018-11-01
世界热带农业信息(2018年6期)2018-03-05
学苑创造·A版(2016年9期)2016-10-10
