
石家庄地区新生儿G6PD缺乏症筛查结果及基因突变分析
2021-07-30 05:22封露露李丽欣马翠霞封纪珍李扬
国际生殖健康/计划生育杂志 2021年4期
封露露,李丽欣,马翠霞,封纪珍,李扬
葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase,G6PD)缺乏症系由G6PD基因突变导致酶活性降低所致,其发病常由误食蚕豆而诱发,故又称蚕豆病[1],是一种常见的遗传性溶血性红细胞酶缺陷病,遗传方式为X连锁不完全显性遗传。根据酶活性缺乏程度与临床表现,可将G6PD缺乏症分为Ⅰ~Ⅴ型5类,其溶血的严重程度与剩余酶的功能呈负相关,临床症状也随之逐步减轻[2]。我国是该病的高发区之一,并呈南高北低的分布特点,患病率一般为4%~15%,海南、广东、广西、云南、贵州、四川、台湾和香港等省和地区较高[3],个别地区可高达40%[4]。G6PD缺乏症在临床上表现为新生儿高胆红素血症、蚕豆病、药物诱导性溶血、某些感染性溶血以及非球形红细胞溶血性贫血,严重者可导致新生儿核黄疸,引起死亡或永久性神经系统的损伤[5-6]。本研究采用荧光技术对石家庄地区出生的237 312例新生儿进行筛查,进一步采用高通量测序技术(next generation sequencing,NGS)对疑似患儿进行基因测序,分析G6PD基因突变情况。
1 对象与方法
1.1 研究对象选择2018年9月—2020年11月于石家庄市内各助产机构出生的237 312例新生儿(男性123 363例,女性113 949例),采用荧光法进行G6PD活性筛查,得到筛查阳性患儿142例,确诊114例。其中56例采集静脉血进行基因测序,另外58例患儿家长拒绝基因诊断。筛查前家长均签署知情同意书,此研究经石家庄市妇幼保健院伦理委员会批准实施。
1.2 方法
1.2.1 仪器与试剂仪器:Wallac VICTOR2D荧光计和Wallac DELFIAPlate Shaker微孔板震荡仪,来源于PerkinElmer公司。试剂:G6PD测定试剂盒(荧光法),来源于Labsystems Diagnostics Oy。
1.2.2 G6PD活性筛查采用G6PD测定试剂盒(荧光法)进行G6PD活性的筛查,方法如下:首先用16 mL底物缓冲液溶解底物;在微孔板孔内分别打入直径3 mm的标准品、质控品和待测样本,每孔加入150μL底物溶液,确保血片完全浸透;微孔板覆膜,室温下避光孵育30 min,振速1 150 r/min;加入150μL冷的铜试剂并测量荧光读数(激发光波长355 nm,发射光波长460 nm)。结果判定:低于切值(3.5 U/g Hb)者为可疑阳性,即G6PD活性可能缺乏,需做进一步诊断。
1.2.3 质量控制筛查实验每块反应板均做单孔标准曲线和高值、低值质控,结果均在允许误差范围内方可发出检测报告。连续2年参加国家卫生健康委员会临床检验中心新生儿筛查室间质评,成绩合格。
1.2.4 G6PD基因测序可疑阳性患儿采集静脉血,送至北京迈基诺医学检验所,利用NGS按照Illumina标准化流程进行G6PD全外显子测序。对发现的致病突变在其所在片段上、下游设计引物进行聚合酶链反应(PCR)扩增及一代验证。对患儿父母也采用Sanger法进行目标基因突变验证。对于未见报道的变异,应用蛋白功能预测软件SIFT、PolyPhen_2、MutationTaster、GERP++和REVEL进行蛋白功能预测。严格按照美国医学遗传学与基因组学会(ACMG)发布的变异解读指南进行突变位点的致病性分析。
2 结果
2.1 石家庄地区新生儿G6PD缺乏症的患病率237 312例新生儿中,筛查阳性142例,确诊114例,总患病率为0.05%。114例患儿中男性105例,筛查结果均值(范围)为1.8(0.4~3.7)U/g Hb;女性9例,筛查结果均值(范围)为2.9(1.8~4.0)U/g Hb,见表1。

表1 石家庄地区新生儿G6PD缺乏症男性患儿及女性患儿对比分析
2.2 G6PD基因突变类型分析114例筛查阳性患儿中58例患儿家长拒绝基因测序,56例进行了基因测序,均为G6PD基因杂合突变,共包含19种突变类型,且均为点突变,其中16种已报道,分别为c.1376G>T、c.1024C>T、c.1388G>A、c.871G>A、c.95A>G、c.487G>A、c.1478G>A、c.406C>T、c.577G>A、c.925A>T、c.185 A>G、c.961 G>A、c.482 G>T、c.653 C>T、c.392 G>T、c.1466G>T;3种未见报道,分别为c.1316G>A、c.575G>C、c.1400C>T,见图1;突变频率较高的分别为c.1376G>T(0.25)、c.1024C>T(0.16)、c.1388G>A(0.14),见表2。
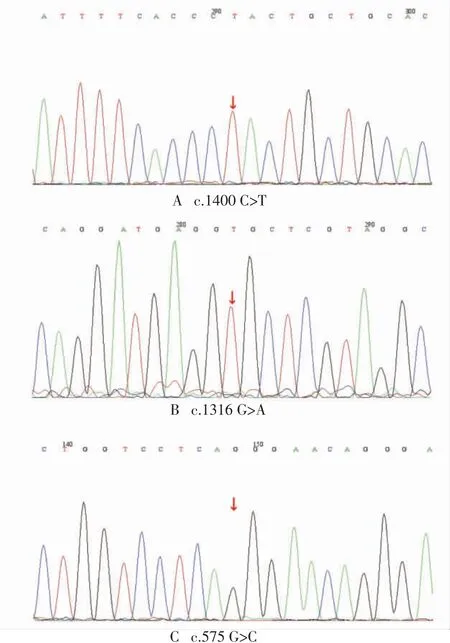
图1 3种未见报道的G6PD基因突变

表2 56例G6PD缺乏症患儿基因突变类型
3 讨论
3.1 G6PD缺乏症的患病率分析G6PD缺乏症在中国儿童总体患病率为4.03%,患病率相对较高,为我国较高的遗传性疾病之一[7]。Chiang等[8]报道中国台湾地区患病率为3.1%~9%;另有报道在广东省进行的一次8 144例筛查中,患病率为3.1%~16.1%[9];云南省患病率为7.39%[10];四川省部分地区患病率达6.9%[11];贵州省的患病率为3.43%[11];香港的患病率为4.4%[12];我国北方地区的山东青岛该病患病率仅为0.02%[13];山东聊城患病率则为1.6%[14];陕西宝鸡地区患病率为0.02%[15]。本研究初步明确G6PD缺乏症在石家庄地区患病率为0.05%,与河北省廊坊市患病率(0.05%)[16]一致,提示G6PD缺乏症在我国发生率存在南高北低的特点,亦表明该病具有种族及地域差异。
3.2 G6PD缺乏症的性别差异G6PD缺乏症是由基因突变引起,致病基因位于X染色体上,故呈现出X连锁遗传的遗传特点。男性半合子酶活性表现为显著缺乏而发病;女性杂合子酶活性呈现不同程度降低,多不发病或轻度发病;女性纯合子会表现出酶活性的严重缺乏[17]。本研究发现56例患儿中除2例女性杂合子,其余均为男性半合子,未发现女性纯合子;男性患病率为0.085%,女性患病率为0.008%。
3.3 G6PD基因突变分析G6PD缺乏症的患病率、基因频率及基因突变类型具有明显的地域和群体特异性。非洲地区主要以c.376A>G、c.202G>A、c.A542T突变为主;地中海地区以及印度等国家主要以c.563C>T为主;东南亚地区则主要以c.871G>A为主;亚洲地区的中国人突变位点主要是c.1388G>A、c.1376G>T、c.95A>G[18-19];本研究表明石家庄地区突变频率较高的位点为c.1376G>T(0.25)、c.1024C>T(0.16)、c.1388G>A(0.14),提示石家庄地区G6PD缺乏症基因突变逐渐呈现地区特征。
综上,本研究通过对石家庄地区新生儿G6PD筛查结果及基因突变分析,明确了G6PD缺乏症在石家庄地区新生儿中的患病率,且男性高于女性;进一步分析了基因突变位点的分布情况;发现了3种未见报道的基因突变类型,丰富了基因突变的数据库。
猜你喜欢
肉类研究(2022年7期)2022-08-05
今日农业(2021年16期)2021-11-26
现代畜牧科技(2021年6期)2021-07-16
现代畜牧科技(2021年6期)2021-07-16
中国生殖健康(2020年2期)2021-01-18
猪业科学(2018年5期)2018-07-17
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
东方艺术·国画(2016年3期)2017-02-08
公民与法治(2016年22期)2016-05-17
