
端环氧基聚醚型聚氨酯预聚体的合成及其弹性体的制备与性能
2021-07-28 07:11卢奕江陈萍萍任浩军
材料科学与工程学报 2021年3期
张 杰,卢奕江,陈萍萍,任浩军,徐 校,杨 康
(华东理工大学 材料科学与工程学院,上海 200237)
1 前 言
聚氨酯(PU)是由柔性链段和刚性链段组成的聚合物,PU具有优异的耐化学性、耐环境性以及较高的机械性能,被广泛用于制造各种材料,如胶黏剂[1-2]、泡沫[3-4]、弹性体[5-6]和涂层材料[7]等,但其在制备过程中通常会使用端NCO的预聚体,从而使其应用受到了限制。这一方面由于PU预聚体的端NCO基团活性较高,易与环境中的水分发生反应,不利于原料储存和产品成型;另一方面,NCO毒性较强,不利于环境和工业卫生。为了解决这些问题,可使用“活性氢”化合物“封闭”或“封端”异氰酸酯基团[8-9],但其解封温度高、耗时长,且封端剂难以除去,从而会对聚氨酯的应用产生不利影响[10-11]。除此之外,PU预聚体的NCO基团还可以用活性氢环氧化合物来封端,得到端环氧基聚氨酯预聚体,从而改善了PU的水敏性和固化成型性[12]。
CHEN等[13]首次以缩水甘油对端NCO聚氨酯预聚体进行封端,得到了端环氧基聚氨酯预聚体(EPU),发现这种类型的聚氨酯的储存稳定性增加,而且还具有与环氧树脂的加工方式相同的优点。YEGANEH等[14]以聚醚(PTMEG、PEG)、异氰酸酯(MDI、HDI)和缩水甘油为原料,制备了环氧聚氨酯弹性体。研究结果表明,PTMEG基的环氧聚氨酯弹性体的机械性能相对较好,当PTMEG分子量为2900时,其拉伸强度可达10.1 MPa,断裂伸长率为377%。MATHEW等[15]通过改变蓖麻油、H12MDI和缩水甘油之间的摩尔比,合成了环氧封端的生物基聚氨酯,研究结果表明,随着摩尔比的增加(1∶3∶3~1∶6∶6),EPU的杨氏模量得到提升,拉伸强度从7.2 MPa上升至10.4 MPa,但其断裂伸长率从55%下降至13%。孙卫红等[16]制备了EPU及EPU/EP的弹性体,研究结果表明,EPU的力学性能均小于5 MPa,但随着EPU/EP共混物中EP比例的增加,弹性体的拉伸强度最高可达22 MPa,但断裂伸长率不足40%。BERA等[17]研究了胺官能化的氧化石墨烯(PPD)对缩水甘油封端的聚氨酯(EPU)的物理机械性能的影响,结果发现,PPD的掺杂会提高EPU的热稳定性和机械性能。
尽管在制备环氧封端的聚氨酯方面已经进行了较多的研究工作,但很少有对硬段为TDI的环氧聚氨酯进行结构与性能的系统性研究,同时,尚未见通过封端剂的结构改性来增强弹性体力学性能的相关研究。因此,本研究选用三种不同分子量的聚醚,并用不同链长的环氧化合物作为封端剂,制备了一系列不同结构的环氧聚氨酯,系统研究了弹性体的结构与性能间的关系,建立了软段分子量、封端剂链长与弹性体的结构和性能的关系式。
2 实 验
2.1 原料与试剂
采用聚四氢呋喃醚二醇(PTMEG-1000,-2000,-3000)、甲苯二异氰酸酯(TDI,T-100)、缩水甘油(98%)、二月桂酸二丁基锡(T-12)、间苯二甲胺(m-XDA),均为工业级,以上原料未经处理直接使用。
参照文献[18]合成单环氧基单羟基聚己内酯(GPLC)(如图1),产物为淡黄色透明液体。FT-IR:3443 cm-1(-OH),1729 cm-1(C=O),908 cm-1(epoxy linkages)。1H-NMR (400 MHz, CDCl3):δ 4.38 (dd, J = 12.3, 2.9 Hz, 1H),4.02 (t, J = 6.6 Hz, 1H),3.86 (dd, J = 12.3, 6.4 Hz, 1H),3.58(s, 2H),3.17 (dd, J = 6.1, 3.1 Hz, 1H),2.81 (t, J = 4.5 Hz, 1H),2.61 (dd, J = 4.7, 2.6 Hz, 1H),2.30(dt, J = 25.5, 7.4 Hz, 3H),1.74-1.45(m, 6H),1.43-1.24 (m, 3H)。ESI-MS,m/z:211.1,325.2,439.2 [M-Na]+。GPLC的环氧当量为261.78 g/mol,平均聚合度为1.66。
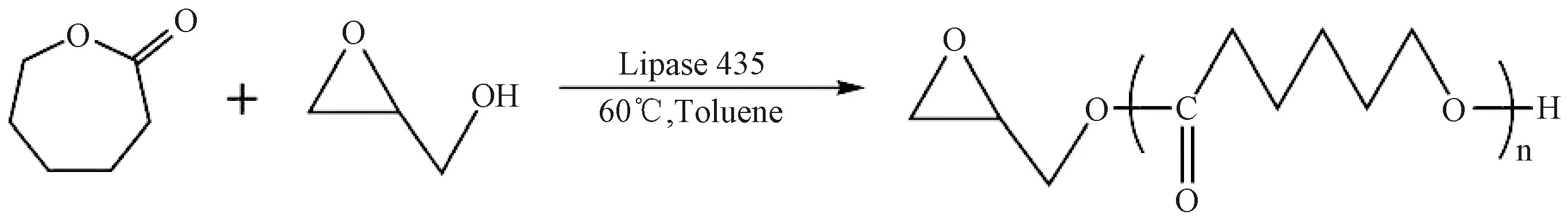
图1 GPLC的合成方程式Fig.1 Synthesis process of GPLC
2.2 端环氧基聚醚型聚氨酯预聚体(EGPU-pre、GGPU-pre)的合成
参考文献[14]合成端环氧聚氨酯预聚体,合成路线如图2所示。在装有搅拌器、温度计的三口烧瓶中加入多元醇,110 ℃下真空脱水2 h,降温至50 ℃以下,加入化学计量的T-100(NCO∶OH=2∶1),缓慢升温至80 ℃反应2~3 h,待NCO含量达到理论值时,停止反应。将物料温度降至50 ℃,并在搅拌下加入化学计量的封端剂(缩水甘油或GPLC,封端剂:TDI∶PTMEG=2.05∶2∶1),缓慢升温至70 ℃,反应2 h,然后加入1滴催化剂T-12,70 ℃下继续反应。当NCO含量低于0.3%时停止反应,得到端环氧基聚氨酯预聚体。预聚体的环氧当量数据和室温状态列于表1中。

图2 端环氧基聚氨酯预聚体的合成路线图Fig.2 Synthesis route of epoxy-terminated polyurethane prepolymer

表1 端环氧基聚氨酯预聚体的环氧当量数据和室温状态Table 1 Epoxy equivalent datas and room temperature state of epoxy-terminated polyurethane prepolymers
2.3 弹性体样品的制备
将上述合成的一定量预聚体置于烧杯中,加入化学计量的m-XDA(epoxy∶N-H=1∶1),混合均匀,脱泡。将得到的树脂倒在离型纸上,用刮棒刮涂,膜的厚度为0.25 mm。将样品置于烘箱中固化,固化条件为80 ℃下加热2 h,100 ℃下后固化4 h,然后于烘箱中自然冷却至室温。另外,撕裂强度的试样于四氟模具中成膜,膜厚约为2 mm。固化样品的结构参数见表2,环氧聚氨酯的结构示意图如图3所示。

表2 弹性体样品的结构参数Table 2 Structural parameters of the elastomer samples

图3 环氧聚氨酯的结构示意图(彩色图)Fig.3 Schematic diagram of structure of epoxy polyurethane
采用ATR FTIR对所制备的弹性体样品进行表征,结果表明,样品谱图中的环氧吸收峰(910 cm-1处)消失,说明体系中的环氧基团已完全反应。
2.4 测试与表征
Nicolet 5700型红外光谱仪(FTIR):扫描32次,扫描范围为500~4000 cm-1,分辨率为4 cm-1;Micromass LCTTM型液相色谱/飞行时间质谱联用仪:配以Waters 600液相色谱进样系统,注射泵离子化方式为电喷雾电离(ESI);AVANCE 400MHz型核磁共振波谱仪(1H-NMR):以氘代氯仿(CDCl3)做溶剂,四甲基硅烷(TMS)为内标;DSC 8500型差示扫描量热仪:在氮气气氛下进行,温度范围-90~150 ℃,升温速率为10 ℃/min,降温速率为20 ℃/min,样品重量为5~10 mg;TGA Q50型热重分析仪(TG):在氮气氛下进行,温度范围为室温~700 ℃,升温速率为10 ℃/min,样品重量为5~8 mg;RINT2000型X射线衍射仪(XRD):25 ℃下测定,测定电压40 kV,电流100 mA,入射角范围5~90°,波长0.154 nm,扫描速度4°/min;Nanoscope IIIa Multimode型原子力显微镜(AFM):探针为硅探针,室温下以中等强度敲击模式(回复率Rsp为0.5)。
力学性能测试按照GB/T 528-2009标准进行,使用AGS-X电子万能试验机在25 ℃下测试,拉伸速率为50 mm/min,样品尺寸为50 mm×4 mm×0.25 mm的哑铃形样品;撕裂强度测试按GB/T 12829-2006标准进行,拉伸速率为500 mm/min;使用邵氏硬度计测量样品硬度。
凝胶含量采用溶剂萃取法测量,以丁酮为溶剂,连续萃取24 h;交联密度采用平衡溶胀法[19-20]测量;环氧值的测量采用盐酸-二氧六环法;吸水率根据国标GB/T 1034-2008进行测量。
3 结果与讨论
3.1 端环氧基聚醚型聚氨酯的合成
通过FTIR测试对端环氧基聚醚型聚氨酯预聚体进行结构表征。图4为原料和产物的FT-IR谱图,可观察到产物谱图中3460 cm-1处的游离羟基峰显著减弱,2270 cm-1处的-NCO吸收峰消失;同时,出现了3340 cm-1处N-H的伸缩振动,1730 cm-1处C=O的吸收峰,1530 cm-1处C-N拉伸与N-H面外弯曲相结合的红外吸收峰,以及1220 cm-1处偶合的C-N和C-O伸缩振动特征峰,表明封端剂中的-OH与预聚体中的-NCO发生了反应,-NCO因完全反应而消失,并生成了氨基甲酸酯键。另外,产物的谱图在910 cm-1处显示了环氧基团的特征吸收峰,表明成功合成了端环氧基聚氨酯预聚体。

图4 原料和预聚体的红外谱图Fig.4 FTIR spectra of raw materials and prepolymers
3.2 聚醚型环氧聚氨酯弹性体的结构表征
3.2.1聚醚型环氧聚氨酯弹性体的交联密度 端环氧基聚氨酯预聚体的端基为环氧基团,使用间苯二甲胺对其进行固化,得到网状交联结构的弹性体材料。为了研究材料的交联密度对性能的影响,使用平衡溶胀法测定材料的交联密度以及交联点间的平均分子质量。根据Flory-Rehner公式,设样品在溶剂中溶胀前后的体积比为Q,VP为Q的倒数,计算公式如式(1)、(2)所示:
(1)
(2)
式中:Wp是聚合物的干重,Ws是样品中溶胀的溶剂重量,ρp是聚合物的密度,ρs是溶剂的密度。
溶剂与样品的相互作用参数χ12可由式(3)计算:
(3)
式中:β是晶格参数,值为0.34;Vm为所用溶剂的摩尔体积,δp是聚合物的溶解度参数,δs是溶剂的溶解度参数,R是气体常数,T是绝对温度。
聚合物的交联点间平均分子质量Mc及交联密度Vc可根据式(4)、(5)计算:
(4)
(5)
通过测定交联样品在8种不同溶剂中的溶胀指数,再以溶胀指数对相应溶剂的溶度参数(表3)作图,然后进行高斯拟合得到拟合曲线(如图5所示),拟合曲线峰值所对应的横坐标,即可求得聚合物样品的溶解度参数δp。交联聚合物样品的密度ρp、Mc和交联密度测量结果列于表4中。

表3 不同溶剂的溶解度参数Table 3 Solubility parameters of different solvents

表4 EGPU和GGPU的交联密度参数Table 4 Crosslinking density parameters of EGPU and GGPU

图5 EGPU(a)和GGPU(b)的溶胀度-溶解度参数的拟合曲线Fig.5 Fitting curves of swelling-solubility parameter of the EGPU (a) and GGPU (b)
由表4中数据可知,随着聚醚和封端剂的分子链长度的增加,弹性体的交联密度下降。这是因为预聚体中的环氧基团是主要的化学交联位点,环氧基团的含量会随着软段分子量和封端剂链长的增加而下降,从而导致体系的交联密度下降。
3.2.2聚醚型环氧聚氨酯弹性体的微相分离 氢键的形成极大地促进了聚氨酯的微相分离,而红外光谱已普遍用于表征聚氨酯内部形成的不同类型的氢键,并由此推断其相分离的结构[21]。因此,为了研究环氧聚氨酯的微相分离,首先通过ATR FTIR研究了EGPU和GGPU的氢键化程度。
图6为EGPU、GGPU羰基区的ATR FTIR谱图,对其进行分峰拟合处理,结果如图7、8所示。图中的1691~1710 cm-1处吸收峰归属于有序氢键化的羰基峰,对应于硬段有序相中氢键结合的羰基;1711~1720 cm-1处的吸收峰归属于无序氢键化的羰基峰,对应于与界面区域或“溶解”在软相中的氨基甲酸酯键所形成氢键的羰基;1721~1740 cm-1处的吸收峰归属于游离羰基的吸收峰[22-23]。表5、表6分别为EGPU、GGPU羰基区的ATR FTIR谱图最小二乘法数据拟合结果,其中X0为氨酯羰基的有序氢键化程度,Xd为无序氢键化程度,Xb为总氢键化程度,R值为硬链段溶解在软链段中的含量,均用于表征聚氨酯中的微相分离程度;f为硬段含量。上述这些系数X0,Xd,Xb,R的计算公式分别为:
(6)
(7)
(8)
(9)

图6 EGPU和GGPU羰基区的ATR FT-IR谱图Fig.6 Carbonyl region of ATR FT-IR spectra for EGPU and GGPU

图7 EGPU1(a)、EGPU2(b)和EGPU3(c)的ATR FT-IR谱图羰基区拟合曲线Fig.7 Fitting curve of carbonyl region of ATR FT-IR spectra for EGPU(a), EGPU2(b) and EGPU3(c)

图8 GGPU1(a)、GGPU2(b)和GGPU3(c)的ATR FT-IR谱图羰基区拟合曲线Fig.8 Fitting curve of carbonyl region of ATR FT-IR spectra for GGPU(a), GGPU2(b) and GGPU3(c)
表5中数据显示,随着软段分子量的增加,EGPU的有序氢键化程度和总氢键化程度下降,且微相分离程度下降,这是由于硬段含量的降低所致。此外,EGPU体系中羰基的有序化氢键程度较低,这可能是由于封端剂缩水甘油的短链结构,使得环氧聚氨酯体系中的硬段与环氧基团紧密相连,环氧的交联结构导致聚氨酯硬段的聚集态结构发生了变化,硬段的有序排列程度降低。为此,本研究希望通过延长封端剂的链长,使硬链段略微远离环氧交联点,减少环氧交联对硬段微相分离和规整排列的影响,从而提升环氧聚氨酯弹性体的微相分离程度以及硬链段的排列规整程度。
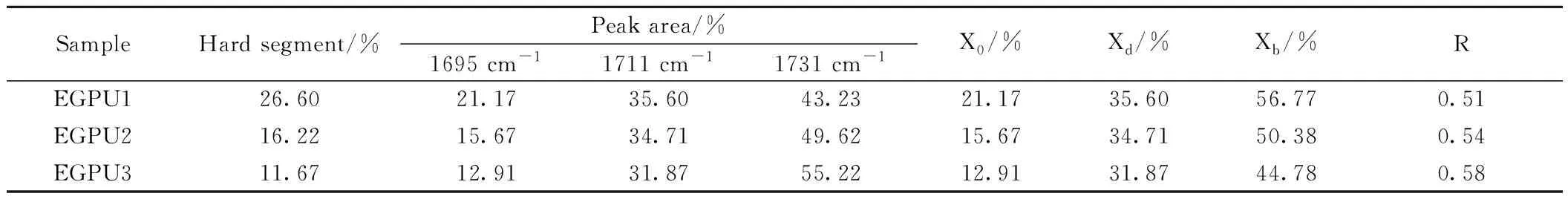
表5 EGPU羰基区的ATR FT-IR谱图最小二乘法拟合数据结果Table 5 Results of Least-square-curve-fitting ATR FT-IR spectra in urethane carbonyl region for EGPU
然而,由于GGPU的大分子链中同时包含了酯羰基(来源于GPLC)和氨酯羰基,其氢键化行为较为复杂,因此简单地用上述公式计算GGPU的氢键化程度,误差较大。为此引入了氨酯羰基贡献因子δ,定义为GGPU的大分子链中氨酯羰基摩尔数与全部羰基摩尔数之比。按式(10)将X0修正为:
(10)
对比表5和表6中数据可以看出,在软段分子量相同的情况下,与EGPU相比,GGPU具有更高的有序氢键化程度以及微相分离程度。这证实了上述的猜想,即增加封端剂的链长,有利于提高环氧聚氨酯中硬段排列的规整程度,从而提高了其微相分离程度。此外,通过DSC和WAXD进一步对EGPU和GGPU的微观形态进行了表征。

表6 GGPU羰基区的ATR FT-IR谱图最小二乘法拟合数据结果Table 6 Results of Least-square-curve-fitting ATR FT-IR spectra in urethane carbonyl region for GGPU
图9为EGPU和GGPU的WAXD衍射图,图中EGPU和GGPU均显示为宽高斯峰,表明所合成的聚醚型端环氧基聚氨酯为无定形态,其软段表现为短程有序排列,且在使用温度(25 ℃)下不结晶。

图9 EGPU和GGPU的WAXD衍射图Fig.9 WAXD diffractograms of EGPU and GGPU
图10为EGPU和GGPU的DSC二次升温曲线,DSC测试结果列于表7中。由图10可观察到,EGPU3和GGPU3的DSC曲线中存在冷结晶峰和结晶熔融峰,结晶熔融温度分别为14.10 ℃、18.41 ℃,表明其在低温下具有良好的结晶性,但在使用温度(25 ℃)下难以结晶,因此在图9中没有观察到软段的结晶衍射峰。

图10 EGPU(a)和GGPU(b)的DSC二次升温曲线Fig.10 DSC secondary heating curves of EGPU (a) and GGPU (b)

表7 EGPU和GGPU的DSC测试结果Table 7 DSC results of EGPU and GGPU samples
在DSC二次升温曲线中均只观察到软段的Tg。由表7中数据可以看出,随着软段分子量的增加,环氧聚氨酯的Tg向低温区移动且较接近于纯软段的Tg,这是由于体系中的连续相软段的含量增加所致,同时也表明所制备的聚醚型环氧聚氨酯具有较好的微相分离程度。此外,在相同软段分子量下,GGPU的Tg略低于EGPU,表明GGPU具有更高的微相分离程度,这与红外分峰拟合的结果一致。
3.2.3聚醚型环氧聚氨酯弹性体的微观形貌 为进一步了解EGPU内部相分离的结构,使用原子力显微镜(AFM)对EGPU1薄膜进行了表征。
图11是EGPU1的AFM高度图(a)、相图(b)和三维图(c)。图11(b)中的亮区对应于高度图(a)中的突起区域,为聚氨酯的硬段区域,而相图中和高度图中的暗色连续相为聚氨酯的软段区域。从(b)图和(c)图中,可以清楚地看到EGPU1样品内软、硬段微区的相分离情况。

图11 EGPU1的AFM高度图(a)、相图(b)和三维图(c)(在室温下以中等强度敲击模式Rsp=0.5测试,扫描范围2 μm×2 μm)Fig.11 AFM tapping mode height image(a), phase image(b) and 3D image(c) of EGPU1 obtained from moderate tapping force at room temperature in air. The scan area is 2 μm×2 μm
3.3 聚醚型环氧聚氨酯弹性体的机械性能
图12为EGPU和GGPU薄膜的应力-应变曲线。从图可见,EGPU和GGPU表现出类似橡胶(软而韧)材料的应力-应变曲线,其力学性能、硬度和凝胶含量数据列于表8中。

图12 EGPU(a)和GGPU(b)薄膜的应力-应变曲线Fig.12 Stress-strain curves of EGPU (a) and GGPU (b) films

表8 EGPU和GGPU的机械性能和凝胶含量Table 8 Mechanical properties and gel content dates of EGPU and GGPU samples
由表中可以看出,当封端剂结构相同时,随着软段分子量的增加,弹性体的断裂伸长率上升,但其撕裂强度和硬度均随之下降,这是由于弹性体的交联密度下降,且聚合物的大分子链柔顺性上升所致。此外,弹性体的拉伸强度表现出随软段分子量的增加而增加的趋势,这可能是由于软链段的拉伸取向结晶并使其应力均匀化所致。因为当PTMEG分子量大于或等于2000的情况下,软段具有结晶倾向,能在高度拉伸时迅速取向成拉伸方向并产生结晶,直到扯断,这一现象在图12的应力-应变曲线中可以观察到,且软段的取向度随拉伸形变的增加而增加[24]。
当软段分子量相同时,封端剂链长的增加会提高聚合物大分子链的柔性,从而导致弹性体的断裂伸长率上升,但其100%定伸应力、硬度和撕裂强度下降。同时,可以观察到弹性体的拉伸强度会随封端剂链长的增加而上升,这是由于环氧聚氨酯的微相分离程度以及酯基含量增加所致。与EGPU3相比,GGPU3的拉伸强度提高了49.17%,断裂伸长率提高了29.33%,提升效果显著。
3.4 聚醚型环氧聚氨酯弹性体的耐热性分析
图13为EGPU和GGPU在N2氛围下的TGA曲线,其TGA测试结果列于表9中。

图13 EGPU(a)和GGPU(b)的TGA测试结果曲线Fig.13 TGA curves of EGPU (a) and GGPU (b)

表9 EGPU和GGPU的TGA测试结果Table 9 TGA results of EGPU and GGPU samples
从图13中可观察到环氧聚氨酯主要表现出两个不同的热降解阶段,第一阶段发生在约250 ℃,为氨基甲酸酯键(聚氨酯硬段)的分解;第二阶段发生在约330 ℃,该阶段涉及聚醚多元醇链段(聚氨酯软段)的分解。由表9中数据可以看出,EGPU和GGPU的热稳定性没有太大差异,且都随着软段分子量的增加而增加,这是由于硬段(热不稳定部分)含量下降,软段(热稳定部分)含量上升所致。
3.5 环氧聚氨酯弹性体的吸水率
亲水性对聚合物的水解具有实质性影响。为此,通过测定吸水率来确定聚合物本体的亲水性,表10列出了环氧聚氨酯弹性体的吸水率测试结果。由表中数据可发现,随着软段分子量的增加,环氧聚氨酯弹性体的吸水率下降,这是因为聚醚链段中碳原子数的增加导致材料表面的亲水性下降。此外,在相同软段分子量下,表现出的吸水率为EGPU>GGPU,这是由于EGPU预聚体中的环氧基团含量更高,而环氧端基与胺的开环反应会产生的额外羟基,从而导致样品的亲水性增加。

表10 环氧聚氨酯弹性体的吸水率Table 10 Water absorption datas for epoxy-terminated polyurethane elastomer samples
4 结 论
1.分别以缩水甘油和GPLC为封端剂,制备了不同结构的聚醚型环氧聚氨酯EGPU和GGPU,所制备的弹性体均具有微相分离的微观形态,且其有序氢键化程度和微相分离程度均随封端剂链长的增加而上升。
2.软段分子量的增加,会提高环氧聚氨酯弹性体的拉伸性能和热稳定性,但会降低其交联密度,导致弹性体的撕裂强度下降。
3.封端剂链长的增加对环氧聚氨酯弹性体的拉伸性能有明显的提升效果,其中与EGPU3相比,GGPU3的拉伸强度提高了49.17%,断裂伸长率提高了29.33%。
猜你喜欢
重庆交通大学学报(自然科学版)(2022年1期)2022-02-10
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
黑龙江生态工程职业学院学报(2020年3期)2020-06-02
装备环境工程(2020年2期)2020-03-23
固体火箭技术(2019年4期)2019-09-13
粘接(2017年7期)2017-08-08
中国塑料(2015年8期)2015-10-14
橡胶科技(2015年1期)2015-02-25
华南理工大学学报(自然科学版)(2014年7期)2014-08-16
中国塑料(2012年6期)2012-12-01
