
基于mRNA生物信息学分析的肺腺癌免疫基因预后风险模型建立与评估*
2021-07-27 06:15葛美玲胡月丁杰刘艳红高玒
临床检验杂志 2021年6期
葛美玲,胡月,丁杰,刘艳红,高玒
(南京大学医学院附属鼓楼医院,a.生物样本库,b.病理科,南京 210008)
在过去的数十年里,肺癌发病率和死亡率逐年升高,目前已经成为癌症相关死亡的主要原因之一。据WHO最新数据显示,肺癌的发病率(11.6%)和死亡率(18.4%)在所有恶性肿瘤中位居第一[1]。在我国,按发病人数顺位排序,肺癌也位于恶性肿瘤发病首位[2]。肺腺癌(LUAD)作为非小细胞肺癌(NSCLC)最常见的组织学亚型,占肺癌发病率的40%。越来越多的证据表明,LUAD发生和发展与肿瘤免疫逃逸密切相关。肿瘤细胞不仅能够通过表达抑制性配体与细胞毒性T细胞表面受体结合抑制其功能,还能通过分泌免疫抑制因子,调节辅助淋巴细胞降低机体的抗肿瘤反应[3]。因此,寻找特异性的免疫分子标志物,开发有效的肿瘤免疫监控手段,探索肿瘤免疫治疗的新方案,对于LUAD患者的治疗及预后具有重要的意义[4]。本研究从分子水平出发,筛选有效的免疫基因标志物,构建预后风险模型,探索免疫基因在LUAD诊断、治疗及预后中的重要作用,并为寻找新的检测手段和治疗靶标提供依据。
1 资料与方法
1.1患者及临床样本
1.1.1患者入组 随机选取2019年5月至9月在南京鼓楼医院进行手术的38例早期LUAD患者。患者手术前均未接受消融、化疗或放疗。患者人口学资料:男性23例(60.53%),女性15例(39.47%),年龄(61.29±10.06)岁。收集上述患者的肿瘤及正常石蜡组织样本76份。
1.1.2病理学评估 将1.1中的石蜡组织制作病理切片,由2位病理科医师检查所有HE染色切片,按照2017年IASLC(国际肺癌研究协会)第八版TNM分期标准及2015年WHO肺腺癌分型标准(贴壁型、腺泡型、乳头型、微乳头型、实体型)对样本进行分期及分型。鉴于肺腺癌的高度异质性,以肿瘤中百分比最高的成分判定主要组织学分型。患者临床病理资料如下:原位肺腺癌3例(7.89%),浸润性肺腺癌35例(92.11%);浸润性肺癌病理分型:贴壁型20例(57.14%),腺泡型10例(28.57%),乳头型3例(8.57%),实体型2例(5.71%)。肿瘤分期:0期3例(7.89%),Ⅰ期35例(92.11%)。肿瘤分化程度:低分化2例(5.26%),中分化17例(44.74%),高分化19例(50.00%)。
1.1.3临床样本全转录组mRNA高通量测序 对1.1.1中的肿瘤组织及其对应的正常肺组织的石蜡标本进行全转录组mRNA高通量测序。样本符合以下要求:(1)病理分期为0期或Ⅰ期;(2)FFPE样本的肿瘤阳性细胞比例>80%。采用测序数据的FPKM值衡量基因的表达量并进行后续分析。
1.2训练集及测试集数据
1.2.1全转录组mRNA数据下载及预处理 从TCGA (https://portal.gdc.cancer.gov/)中下载肺腺癌(LUAD)的mRNA测序数据(HTSeq-FPKM)及临床数据作为训练集进行后续分析。在GEO平台中下载数据集GSE101929、GSE50081、GSE41271、GSE42127 mRNA测序数据及临床数据作为测试集,使用svaR软件包矫正各数据集测序数据的多批次效应,将得到的结果用于后续分析。
1.2.2患者临床数据筛选 选取截至2020年6月收录于TCGA数据库以及GEO数据库中GSE101929、GSE50081、GSE41271、GSE42127数据集中的LUAD患者。纳入标准:(1)病理诊断为LUAD,且无其他恶性肿瘤;(2)mRNA测序数据及随访资料完整,随访天数大于30 d。(3)同例患者存在不同类型肿瘤样本时,仅选择1个肿瘤样本的数据。不同类型样本选择顺序:冰冻组织>石蜡组织。本研究中训练集共纳入患者439例,测试集共纳入患者476例。
1.3肺腺癌预后模型的建立及验证
1.3.1训练集中肿瘤及正常样本差异表达基因分析 对训练集mRNA测序数据进行预处理,利用limma R[5]软件包从训练集中鉴定了439例肿瘤组织样本及52例正常肺组织样本之间的差异表达基因(differently expressed genes,DEGs)。以log2-fold change (|FC|)>1和错误发现率(false discovery rate,FDR)<0.05作为阈值。
1.3.2预后模型的建立及验证 将肿瘤与正常样本的DEGs与ImmPort数据库(https://www.immport.org/home)提供的免疫相关基因取交集筛选出候选基因。采用单变量COX回归对候选基因与患者总生存时间(OS)进行分析,筛选P<0.05的基因。然后采用Lasso回归筛选上述基因并构建模型[6-7],计算调整参数lambda值(tuning parameter,λ)。通过识别与交叉检验误差均值最小值对应的λ值作为依据,筛选变量计算回归系数,构建预后风险模型[8]。由回归模型得到的回归系数乘以其mRNA水平产生了每个样本的预后风险评分(risk score)。选择训练集的风险评分中位值作为阈值进行分组,将风险分值大于阈值的患者归为高危组(high risk group),分值小于或等于阈值的患者归为低危组(low risk group)。采用相同的公式和阈值计算测试集中患者的风险评分并进行分组。分别绘制2个队列患者的生存曲线并计算高、低危组患者OS的差异。分别绘制两队列患者的总生存时间的ROC曲线并计算曲线下面积(area under curve,AUCROC),评估风险模型的预测性能[9]。
1.4统计学分析 所有统计分析均使用R软件(4.0.2)进行。使用Kaplan-Meier法进行生存分析并绘制曲线。采用Log-rank检验(Log-rank)分析生存差异的统计学意义。采用LASSO回归构建预后风险模型。通过ROC曲线评价预后模型的效能。采用卡方检验(Chi-square test)或Fisher精确检验(Fisher′s exact test)分析高、低危组间的临床病理资料。所有统计检验均为双侧检验,以P<0.05为差异有统计学意义。
2 结果
2.1基于训练集的肺腺癌预后模型构建 对训练集数据中肿瘤与正常组织mRNA测序数据进行比较,筛选出12 946个DEGs进行下一步分析。其中,与正常组织相比,10 207个DEGs在肿瘤组织中被上调,2 739个DEGs被下调。将这些DEGs与免疫基因进行交集筛选出1 091个基因。采用单因素COX 回归分析这些基因与OS的相关性,筛选出86个候选基因(P<0.05)。将上述候选基因纳入LASSO回归分析,并计算最优λ值。根据最优λ值重新拟合数据,筛选出12个基因包括:纤维母细胞生长因子2(fibroblast growth factor 2,FGF2)、轴突导向因子7A(semaphorin 7A,SEMA7A)、抑制素亚基α (inhibin subunit alpha,INHA)、葡萄糖-6-磷酸异构酶 (glucose-6-phosphate isomerase,GPI)、血管生成素样蛋白4(angiopoietin like 4,ANGPTL4)、肿瘤坏死因子受体超家族成员11 A (TNF receptor superfamily member 11a ,TNFRSF11A)、细胞色素b-245 β链 (cytochrome b-245 beta chain,CYBB)、精氨酸酶2 (arginase 2,ARG2)、尾加压素2 (urotensin 2,UTS2)、白血病抑制因子受体亚基α (LIF receptor subunit alpha,LIFR)、SHC适配器蛋白3(SHC adaptor protein 3,SHC3)、细胞毒性T淋巴球相关蛋白4(cytotoxic T-lymphocyte associated protein 4, CTLA4),然后计算LASSO回归系数构建公式(图1A、B)。公式如下:风险评分= (-0.001 77×CYBBmRNA水平)+(0.170 269×FGF2mRNA水平)+(-0.045 58×ARG2mRNA水平)+(0.0051 21×SEMA7AmRNA水平)+(0.005 625×GPImRNA水平)+(0.003 913×INHAmRNA水平)+(-0.133 98×UTS2mRNA水平)+(0.043 29×ANGPTL4mRNA水平)+(-0.016 18×LIFRmRNA水平)+(0.144 495×TNFRSF11AmRNA水平)+(-0.083 3×SHC3mRNA水平)+(-0.043 53×CTLA4mRNA水平)。

注:A,Lasso回归分析中的调节参数(λ)与模型误差图。横坐标为λ的对数值,纵坐标为模型误差,图形最上方为模型筛选出来的对应λ的变量个数。以最小二项偏差筛选出12个变量; B,Lasso 回归系数路径图。图中的每条线代表了1个变量,纵轴为回归系数,横坐标为λ的对数值。10倍交叉验证法对log(λ)进行测试筛选出12个非零的变量。
LASSO回归中FGF2、SEMA7A、INHA、GPI、ANGPTL4、TNFRSF11A的正系数提示其高表达与较差的OS有关,而CYBB、ARG2、UTS2、LIFR、SHC3、CTLA4的负系数提示其高表达与较好的OS有关。根据中位风险评分(0.334),将训练集中所有患者分为高危组(221例)和低危组(218例)。生存分析显示低危组患者3年、5年生存率分别为80.2%和62.5%,高危组患者3年、5年生存率分别为44.2%和23.7%。高危组的总生存率明显少于低危组患者(P<0.001,图2A~C)。将所有患者的12个基因表达谱可视化为热图(图2D)。训练集的预后模型AUCROC为0.759 (图2E)。表明该模型在生存监测中具有良好的效力。基于自变量分析建立了预测1年、2年、3年生存率的列线图,年龄、病理分期(stage)、肿瘤大小分期(T)、淋巴结转移(N)、远处转移(M)及风险评分(risk score)均作为参数纳入列线图(图2F)。

注:A,高危组及低危组患者OS比较; B,高危组及低危组患者的风险曲线; C,高危组及低危组患者的生存状况; D,高危组及低危组患者12个模型基因表达热图; E,以训练集中患者年龄、性别、病理分期、肿瘤大小、淋巴结转移、远处转移、风险评分分别作为变量的生存预测模型的ROC曲线; F,以训练集中患者性别、病理分期、肿瘤大小、淋巴结转移、远处转移、风险评分分别作为变量预测患者1年、2年、3年生存率的列线图。
2.2基于测试集的肺腺癌预后模型验证 在测试集中评估预后风险模型的预后预测性能,使用上述风险评分公式计算测试集患者的风险评分,使用与训练集患者风险评分相同的阈值按同样的方式将测试集划分为高危组(122例)和低危组(354例)。在两组人群的生存分析中,低危组患者3年、5年总生存率分别为72.8%和59.3%,高危组患者3年、5年总生存率分别为53.7%和42.5%,高危组的总生存率明显低于低危组(P<0.001,图3A~C)。将测试集中所有患者的12个模型基因表达谱可视化为热图(图3D)。模型 AUCROC为 0.707(图3E)。结果表明,预后模型对预测测试集患者生存预后的表现较好。基于自变量分析建立了预测患者1年、2年、3年生存率的列线图,年龄、病理分期、风险评分均作为参数被纳入列线图 (图3F)。

注:A,高危组及低危组患者OS比较; B,高危组及低危组患者的风险曲线;C,高危组及低危组患者的生存状况;D,高危组及低危组患者中12个基因表达热图; E,以测试集中患者年龄、性别、病理分期、风险评分分别作为变量的预测模型的ROC曲线; F,以测试集中患者年龄、性别、病理分期、风险评分分别作为变量预测患者1年、2年、3年生存率的列线图。
2.3预后风险模型对早期肺腺癌患者预后的预测潜能 对训练集及测试集中肿瘤早期(Ⅰ期)患者的总生存时间与风险评分进行分析,训练集的AUCROC为 0.709(图4A),测试集的 AUCROC为 0.639(图4B)。表明该模型对早期LUAD的预后也有较好的预测潜能。对训练集患者的风险评分与各项临床病理数据进行分析,该评分随着患者Stage(图5A)和T分期(图5B)进展逐渐升高,Ⅰ期患者肿瘤样本的风险评分明显高于正常组织(P<0.05),淋巴结转移患者的得分明显高于未转移患者(P<0.05,图5C),但远处转移患者与未转移患者之间的风险评分差异无统计学意义(P>0.05,图5D)。

注:A,训练集中患者年龄、性别、病理分期、肿瘤大小、淋巴结转移、远处转移及风险评分分别作为变量的生存预测模型的ROC曲线;B,以测试集中早期LUAD(Ⅰ期)患者年龄、性别、病理分期、风险评分分别作为变量的生存预测模型ROC曲线。
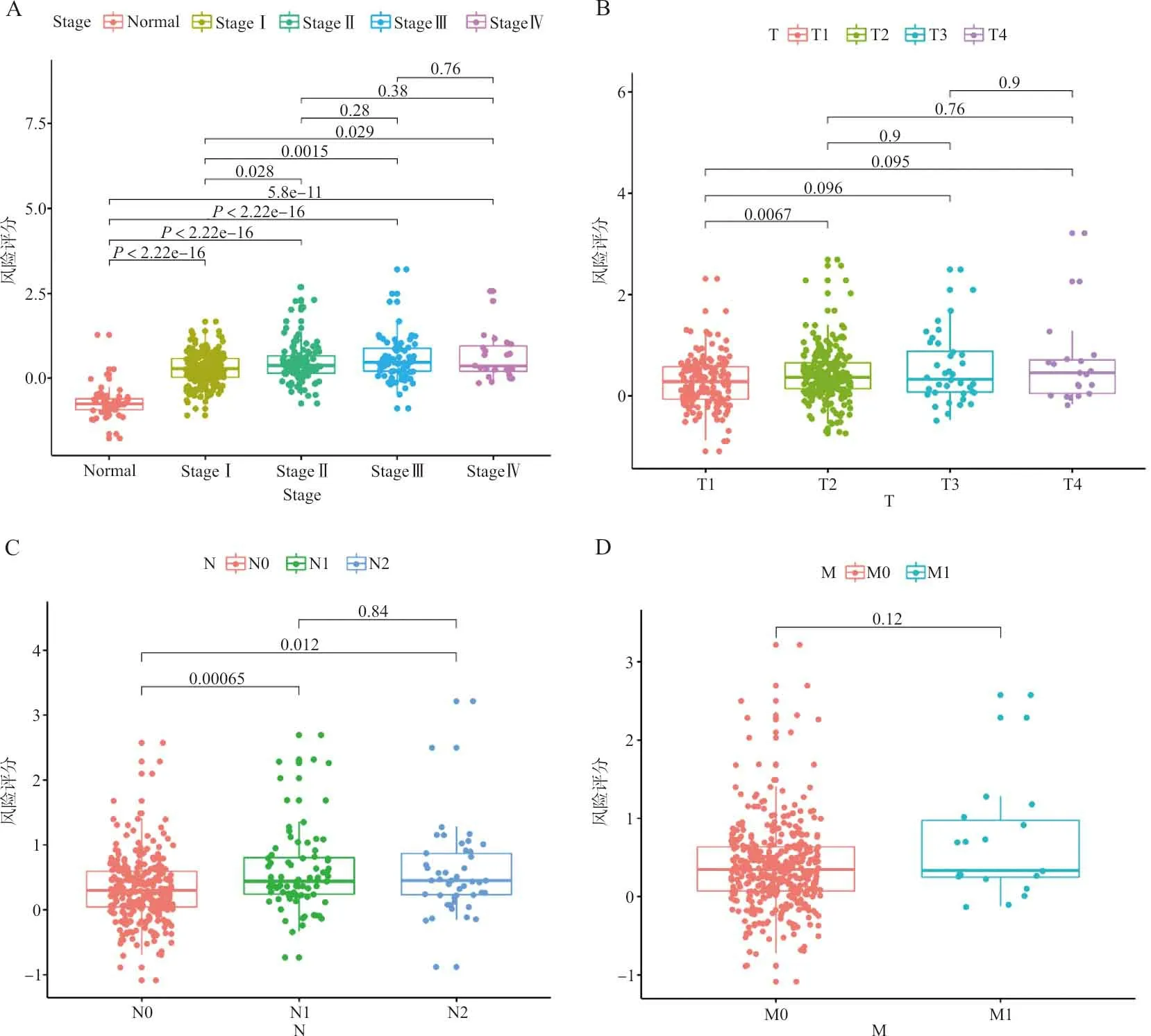
注:A,正常样本及肿瘤Ⅰ、Ⅱ、Ⅲ、Ⅳ期样本的风险评分比较; B,不同肿瘤大小分期(T1、T2、T3、T4)患者的风险评分比较; C,未发生淋巴结转移及淋巴结转移患者的风险评分比较; D,未发生远处转移及远处转移患者的风险评分比较。
2.4预后模型与临床早期肺腺癌患者病理参数的相关性 本研究对临床早期肺腺癌患者的转录组测序数据进行了分析,对比38例患者的肿瘤与正常组织mRNA表达情况(图6),CYBB、FGF2、LIFR、SHC3基因在正常组织中的表达量高于肿瘤组织,差异具有统计学意义(P<0.05);GPI、UTS2、TNFRSF11A、CTLA4基因在正常组织中的表达量低于肿瘤组织,差异有统计学意义(P<0.05)。依据上述构建的预后风险模型阈值(0.334),将早期肺腺癌患者分为高危组和低危组。高危组患者12例,低危组患者26例。比较不同预后模型分组患者的临床病理参数差异(表1)。两组患者在性别、年龄、吸烟史、分化程度、TNM分期及肿瘤大小分期方面差异无统计学意义(P>0.05),但高危组患者的肿瘤最大径大于低危组患者,该组乳头型或实体型肺腺癌样本比例也高于低危组,差异均有统计学意义(P=0.029,P=0.044)。
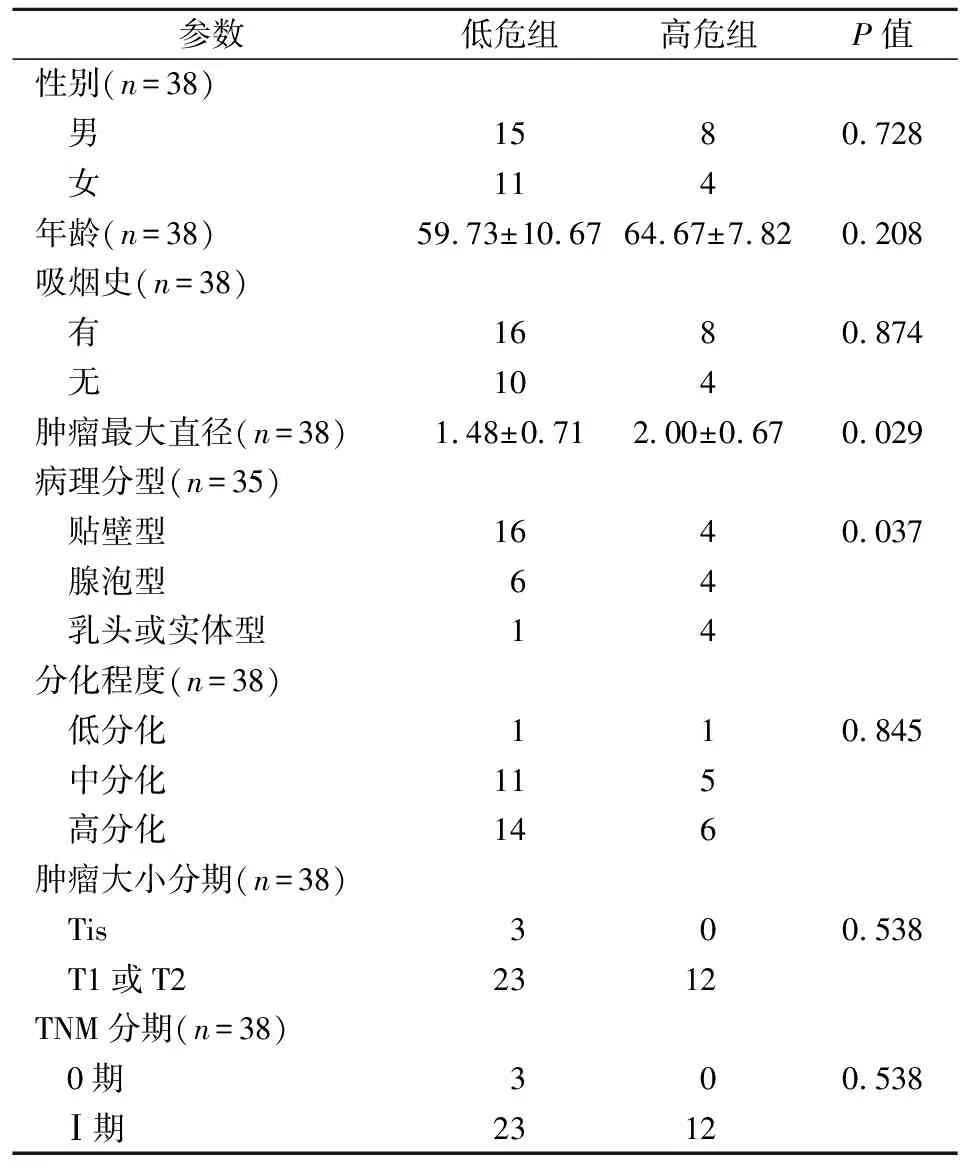
表1 高、低危组患者各项临床病理参数比较

注:纵坐标为基因表达量,取测序数据FPKM值的对数值(Log2);横坐标为12个基因的基因名称。红色图标代表肿瘤组织基因表达量,蓝色图标代表正常组织基因表达量。
3 讨论
大量的研究表明,免疫微环境在肺腺癌的发生、发展中起着重要的作用。探索异常表达的免疫基因对于确定肿瘤发生、发展机制,发现潜在治疗靶点具有重要意义。对于LUAD这种异质性很高的肿瘤而言,单一功能的基因模型很难完整评估所有患者的免疫状态。本研究基于不同来源的数据集,构建了包含不同功能的免疫差异基因的预后模型,试图开发一种方便可靠的方法来监测LUAD患者的免疫状态并评估预后。
本研究建立的模型筛选了12个免疫基因。这些基因涉及不同的蛋白质、通路及功能:CTLA4主要表达在活化的 T 细胞及Treg细胞表面,是第1个被证实的靶向的免疫检查点分子[10],并在抑制机体免疫反应及促进肿瘤免疫逃逸中起着重要作用[11]。FGF2是一种多肽类生长因子,可以激活Ras-Raf-MapK、PI3K/Akt/mTOR等信号通路,促进肿瘤细胞生长、增殖、分化及转移等行为[12]。UTS2是一种生长抑素样环状多肽,能够促进肿瘤血管形成,该基因促新生血管形成的作用与FGF2相似[13]。LIFR是白血病抑制因子的受体,能够介导由白细胞介素6相关因子引起的信号参与免疫调节,并参与PI3K/Akt通路从而影响LUAD发生、发展[14];TNFRSF11A作为肿瘤坏死因子超家族细胞因子,能够抑制肿瘤细胞的能动性和迁移[15]。GPI是细胞内糖酵解和糖异生过程中的关键酶,以自分泌的形式与细胞自身或邻近细胞表面受体相结合[16],影响肺肿瘤细胞的凋亡、迁移及浸润等行为[17]。ARG2可能通过阻碍聚乙二醇化的ARG1与配体结合来抑制后者的抗肿瘤作用[18]。ANGPTL4能够促进肿瘤细胞肺转移[19];SHC3被认为是T细胞受体和B细胞受体信号传导的负调节因子[20];CYBB作为NOX2的亚基,其表达缺失能够减少肿瘤细胞的肺转移[21]。INHA与受体TGFβRⅢ结合能够拮抗激活素,具备抑制多种细胞肿瘤化的效应[22]。信号素蛋白SEMA7A通过调控mTOR信号通路,并被EGFR通路诱导表达,促进肺腺癌发生,可以作为EGFR突变的肺腺癌预测生物标志物及新治疗靶点[23]。
为了进一步探索预后模型在早期肺腺癌中的应用前景,笔者分析了训练集中不同病理分组患者的风险评分,结果发现该评分在早期肿瘤组织中显著高于正常组织,具有成为早期肺腺癌诊断标志物的潜力。生存分析也揭示预后模型对于早期肿瘤患者的生存时间具有一定的预测能力。同时,笔者依照模型基因及参数结合测序数据对38例临床早期肺腺癌患者样本进行了验证,发现与正常肺组织相比,CYBB、FGF2、LIFR、SHC3基因在早癌组织中表达降低,GPI、UTS2、TNFRSF11A、CTLA4基因在早癌组织中表达升高。随后,将患者进行了高、低危分组,并评估了两组患者的临床病理参数的差异,结果发现肿瘤最大径及病理分型在高、低危组患者之间存在差异。高风险评分与更大的肿瘤直径以及特异的病理分型(乳头或实体型)有关。已有研究表明[24],相对于贴壁型肺腺癌患者,实体型为主的肺腺癌患者5年生存率低,预后较差。因此,笔者认为具有高风险评分的早期肺腺癌患者可能预后更差。
综上所述,本研究构建了1个免疫差异基因的预后风险模型,该模型对于肺腺癌患者预后具有良好的预测能力,相关免疫基因可能成为早期肺腺癌诊断的生物标志物。然而,本研究尚存在许多局限性。本研究构建的模型是建立在生物信息及统计学分析基础上的,筛选出的模型基因及其对下游信号通路可能产生的影响还需要生物学实验的进一步验证。虽然笔者对该模型进行了临床验证,发现其对于早期肺腺癌预后有一定的预测能力,但本研究选择的样本量较少而且患者的生存及预后数据尚不完善,这种预测能力的可靠性还需进一步探索。我们希望本研究结果能为肺腺癌患者早期诊断、风险分层、预后评估及免疫治疗提供新的研究方向。
猜你喜欢
延安大学学报(医学科学版)(2022年2期)2022-11-23
临床军医杂志(2022年7期)2022-08-06
岭南心血管病杂志(2022年3期)2022-06-27
中国典型病例大全(2022年12期)2022-05-13
昆明医科大学学报(2022年2期)2022-03-29
检验医学与临床(2022年5期)2022-03-21
昆明医科大学学报(2022年1期)2022-02-28
保健医苑(2021年7期)2021-08-13
现代临床医学(2021年4期)2021-07-31
抗癌(2021年2期)2021-07-09
