
全外显子测序技术鉴定一个常染色体显性遗传性痉挛性截瘫3A型家系ATL1基因突变
2021-07-24 01:53郑备红孙艳邱淑敏陈晓菁杜生荣杨春梅
实用医学杂志 2021年13期
郑备红 孙艳 邱淑敏 陈晓菁 杜生荣 杨春梅
1福建省妇幼保健院,福建医科大学附属医院(福州350001);2福建省生殖医学中心(福州350001)
遗传性痉挛性截瘫(hereditary spastic paraplegias,HSP)是一组具有高度临床和遗传异质性的神经系统变性疾病[1-2],发病率约为1.8/100 000[3]。根据其临床表型,HSP 可分为单纯型和复杂型,单纯型HSP 主要临床特征为渐行性双下肢痉挛性肌无力、肌张力增高、腱反射活跃亢进及痉挛步态;复杂型HSP 除了表现出痉挛性截瘫的症状外,还可伴有其他神经系统的症状,如小脑共济失调、智力迟钝、癫痫、耳聋、视网膜病变、视神经萎缩、肌肉萎缩、皮肤病和周围神经病等[4-6]。HSP 遗传方式有常染色体显性遗传(autosomal dominant,AD),常染色体隐性遗传(autosomal recessive,AR)和X连锁遗传(X-linked,XL),也可见母系遗传(maternal inheritance)(或线粒体相关遗传)[7-8],其中以AD-HSP最为常见。截止目前,已报道的HSP亚型有70 多种,并已鉴定出超过80 个基因与HSP 相关联,根据其基因型被发现的顺序命名为SPG1-77[9-13]。
中国人群AD-HSP 主要以痉挛性截瘫类型4(SPG4)和痉挛性截瘫类型3A(SPG3A)为主[14]。AD-HSP 常见致病基因有SPAST、ATL1、REEP1 和KIF5A 等[13-14]。典型的SPG4 发病年龄都在青少年或成年后发病,并且进展缓慢。而SPG3A 主要表现为单纯型HSP,极少数患者表现为复杂型HSP,该病发病年龄主要集中于儿童时期(<10 岁),ATL1(OMIM182600)基因变异是引起SPG3A 型主要致病原因。本研究应用全外显子测序技术在一个三代遗传性痉挛性截瘫家系中鉴定了一个ATL1 基因杂合错义突变c.715C >T,Sanger 测序验证该突变在家系中表现出基因型与表型共分离,明确致病突变可为家系成员提供准确可靠的遗传咨询和产前诊断。
1 对象与方法
1.1 研究对象本研究家系来自福州(图1),连续三代发病,共4 名患者,其中女性患者2 名,男性患者2名,先证者(Ⅳ:13)为35岁女性,自幼双下肢便开始出现僵硬、活动不便、行走无力,主要症状表现为双下肢肌张力增高、腱反射活跃亢进、病理征阳性,行走呈剪刀步态,且症状随着年龄增长呈进行性加重,能缓慢独立行走,患者智力正常,精神发育正常,余未见异常,符合典型的单纯性痉挛性截瘫。家系中先证者的外公(Ⅱ:1)、母亲(Ⅲ:8)和舅舅(Ⅲ:3)3 名成员皆有类似症状,据先证者自述,其母亲和舅舅自学会走路却出现行走困难的临床表现,家系中其余成员均无相似临床症状,根据家系图谱(图1)可初步推断该病呈常染色体显性遗传模式。本研究中所有参与者均签署了知情同意书,并得到了福建省妇幼保健院伦理委员会的批准。

图1 遗传性痉挛性截瘫家家系图Fig.1 Family diagram of hereditary spastic paraplegia.The arrow indicates the proband
1.2 方法
1.2.1 外周血基因组DNA 提取采集先证者及其家系成员静脉血2 mL,利用DNA 提取试剂盒(Blood Genomic DNA Mini Kit,德国Qiagen 公司)提取基因组DNA,用Nanodrop 2000(美国Thermo 公司)超微量核酸蛋白测定仪对样本DNA 进行质控。使用Qubit 核酸定量仪(美国Thermo 公司)对DNA 浓度进行精确定量。
1.2.2 建库捕获和高通量测序本研究采用安捷伦v6 捕获芯片系统(Agilent SureSelect Human All ExonV6 试剂盒)进行建库和捕获实验,严格按照最新优化的实验步骤操作:首先使用Covaris 破碎仪将基因组DNA 随机打断成长度为180 ~280 bp的片段,片段两端经末端修复和加A 尾后分别连接上接头制备成DNA 文库。带有特异index 的文库pooling 后与生物素标记探针完成液相杂交,再采用带链霉素的磁珠将基因上的外显子捕获下来,PCR 线性扩增后构建文库,经Agilent 2100 生物分析仪器(美国Agilent 公司)对文库质量检测合格后,在Illumina HiSeq 2500 测序仪(美国Illumina 公司)上完成高通量测序。
1.2.3 数据筛选和生物信息学分析目标区域测序原始数据经过质量控制后,应用BWA 软件与人基因组数据库(HGl9)进行比对,采用GATK 和VarScan 软件对单个核苷酸多态位点(SNP)、小片段缺失/插入(InDels)进行分析,重点关注呈常染色体显性遗传的基因,应用dbSNP 数据库、ExAC数据库、HapMap 数据库、千人基因组计划数据库完成数据过滤筛选,保留突变频率小于1%的位点,通过SIFT 和Polyphen-2 等在线功能预测工具对可疑突变位点进行致病性预测,检索HGMD 和Clinvar 等相关基因突变数据库对可疑致病位点进行确认,同时依据美国医学遗传学与基因组学会发布的《序列变异解读标准和指南》对序列变异位点判读,基于先证者表型筛选出有害变异。
1.2.4 Sanger 测序验证采用Sanger 测序方法对全外显子测序分析筛选出的可疑基因致病突变位点进行家系共分离验证。利用premier 5 软件设计引物对gDNA 进行PCR 扩增,正向引物:5′-TGTGAGAAGAGCAGGTTTCAGGAGT-3′,反向引物:5′-TCCTCAAGATAAAAGGGGACAATA-3′。扩增体系及条件,总体系20 μL,包括7.5 μL Q×Buffer,2.0 μL 10×Buffer,1.0 μL MgCl2,四种dNTPs(10 mmol/L)0.4 μL,Hotstar Taq(5 U/L)0.1 μL,上下游引物(100 ng/L)各2 μL,DNA 模板(100 ng/L)1 μL,ddH2O 加至20 μL。反应条件为:95 ℃预变性5 min,95 ℃30 s,62 ℃30 s,72 ℃30 s,32 个循环,最终72 ℃延伸10 min,PCR 产物纯化后经ABI 3170 测序仪(美国ABI 公司)完成双向测序,测序结果与ATL1 基因(转录本序列NM_015915.4)进行比对。
2 结果
通过全外显子测序和生物信息学分析,在数据库中分析比对后发现先证者(Ⅳ:13)ATL1 基因第10 号外显子存在c.715C >T 杂合突变,引起编码蛋白的第239 位氨基酸由精氨酸变成半胱氨酸(p. R239C),该变异位点在dbSNP、ExAC、ESP6500和1000 Genomes 等均未见收录,依据ACMG 判读该位点为疑似致病,既往已有文献报道ATL1 基因c.715C >T 杂合突变与致遗传性痉挛性截瘫3A型相关。采用Sanger 测序方法对先证者及其家系成员gDNA 进行验证,测序结果发现家系中患者(Ⅱ:1,Ⅲ:3,Ⅲ:8)均携带该杂合突变位点,先症者突变位点遗传自母亲,而其他正常成员均未携带此突变,从而证实基因型和疾病表型完全共分离,表明ATL1 基因c.715C >T 为该家系发生遗传性痉挛性截瘫致病原因。
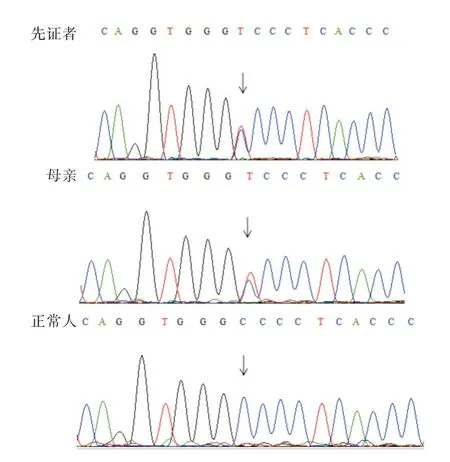
图2 Sanger 测序验证结果Fig.2 Sanger sequencing results
3 讨论
遗传性痉挛性截瘫是一组与神经系统退行性病变相关的单基因遗传病,可发病于任何年龄段,但以儿童和青春期多见[3-4]。由于亚型多且表型复杂,临床上极易误诊漏诊,需与遗传性共济失调、肌萎缩侧索硬化症及肾上腺脊髓神经病相鉴别诊断。所有遗传性痉挛性截瘫类型中,文献报道研究最多的是常染色体显性遗传痉挛性截瘫(AD-HSP),其中以痉挛性截瘫类型3A(SPG3A)、痉挛性截瘫类型4(SPG4)最为常见,分别占40%和10%[13]。
SPG3A 是由ATL1 基因突变导致的单纯性ADHSP,ATL1基因定位于14q22.1,包含14个外显子,编码一个由553 个氨基酸组成的Atlastin-1 蛋白[15]。Atlastin-1 蛋白是一种与动力蛋白相关的GTP 酶,主要表达于脑部组织的皮质、海马和皮层脊髓束神经元,参与管状内质网的形成和轴突的伸长[16]。NAMEKAWA 等[17]发现ATL1 基因变异可造成GTP酶活性缺失,可通过显性负效应抑制内质网的形成及内质网运输囊泡至高尔基体,形态异常的内质网和高尔基体会引起皮质脊髓束等神经元的死亡,从而造成SPG3A 疾病的发生。通常ATL1基因缺陷的患者发病年龄较早,本研究中的患者刚学走路时即表现出行走困难,其临床表型符合SPG3A 型诊断特征。
迄今为止,国内外已报道了ATL1 基因致病突变类型90 余种,绝大多数为错义突变,少数为剪接位点、小片段缺失/插入和大片段缺失突变,错义突变分布较为平均。本研究通过对先证者全外显子组测序检测,发现先证者(Ⅳ:13)ATL1 基因第10 号外显子存在c.715C >T 杂合突变,引起编码蛋白的第239 位氨基酸由精氨酸变成半胱氨酸(p. R239C)。既往已有多篇文献报道该位点与遗传性痉挛性截瘫3A 型相关[18-20],ZHAO 等[21]在对142 个遗传性痉挛性截瘫家系ATL1 基因筛查中发现,绝大多数突变发生在ATL1 基因的第4、7、8 和12 号外显子上,其中有31 个家系均携带c.715C >T(p. R239C)杂合错义突变(21.83%),表明第239位氨基酸是一个高频致病突变位点。
目前尚无有效的针对HSP 的治疗方法,现阶段常见做法是联合药物和物理疗法来缓解HSP 的症状是,一定程度上改善患者的生活质量[22]。药物的作用主要是抑制突触反射力,松弛肌张力及补充神经营养;物理疗法包括推拿按摩、电刺激和针灸等。对于严重的痉挛畸形可采用手术进行矫正,以改善痉挛步态。
综上所述,本研究在一个三代遗传性痉挛性截瘫家系中采用全外显子测序技术鉴定了一个已报道的ATL1 基因c.715C >T(p.R239C)杂合错义突变,明确致病突变可为家系成员提供准确可靠的遗传咨询和产前诊断。HSP 在家系中呈常染色体显性遗传,患者的子女中将有50%的发生风险,且男女患病的机会相等。因此,建议家系中的患者生育时须行产前诊断或植入前遗传学诊断,以阻断遗传病向子代传递。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国肿瘤临床(2022年14期)2022-08-09
基层中医药(2022年1期)2022-07-22
临床输血与检验(2022年3期)2022-06-22
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
中国科技纵横(2020年16期)2020-11-28
云南中医中药杂志(2020年3期)2020-05-11
诊断学(理论与实践)(2020年1期)2020-04-28
健康大视野(2020年6期)2020-04-10
