基于计算机模拟技术以埃博拉病毒跨膜糖蛋白为靶点挖掘中药抗病毒活性分子
2021-07-15 11:26史海龙史永恒刘继平
中草药 2021年13期
史海龙,史永恒,王 川,程 怡,黄 月,刘继平,王 斌
基于计算机模拟技术以埃博拉病毒跨膜糖蛋白为靶点挖掘中药抗病毒活性分子
史海龙,史永恒,王 川,程 怡,黄 月,刘继平,王 斌*
陕西中医药大学,陕西 咸阳 712046
基于埃博拉病毒跨膜糖蛋白(transmembrane glycoprotein of the Ebola virus,EBOV-GP)的三维空间结构,运用计算机虚拟筛选技术从中国台湾中医药数据库TCM@TAIWAN中寻找具有抗病毒作用的中药活性分子。以EBOV-GP为靶点,采用类药性评估、分子对接、分子动力学模拟以及吸收、分布、代谢和排泄(absorption、distribution、metabolism and excretion,ADME)预测等多尺度虚拟筛选策略,从TCM@TAIWAN数据库中挖掘候选药效分子,并分析候选分子与靶点的相互作用情况。以胺碘酮为阳性对照药物,筛选出4个类药性良好的中药单体ZINC85531496、ZINC85567560、ZINC85592968、ZINC33833122,与EBOV-GP亲和力及相互作用基团均优于胺碘酮。采用多种计算机虚拟筛选技术挖掘EBOV-GP抑制剂,可促进从天然产物化学结构数据库中提取、设计以及实验合成抗埃博拉病毒的药效分子。
埃博拉病毒;跨膜糖蛋白;虚拟筛选;中药;抗病毒活性分子
埃博拉病毒是一种致病性致病菌,可以引起人类和动物出血热,并在感染后7~11 d导致90%病例死亡。目前很多药物和疫苗尚处于研发或临床试验阶段,因此迫切需要研发安全性高、耐药性好的抗病毒药物[1]。
埃博拉病毒带有1个负义RNA基因组,包含7个编码不同结构蛋白的基因,分别为、、、、糖蛋白、核蛋白和聚合酶。埃博拉病毒跨膜糖蛋白(transmembrane glycoprotein of the Ebola virus,EBOV-GP)是病毒体表面上唯一在病毒中表达的蛋白,对于附着于宿主细胞和催化膜融合至关重要。因此,EBOV-GP是疫苗的关键组成部分,也是中和抗体以及附着和融合抑制剂的靶点。2016年Pallesen等[2]使用低温冷冻电镜技术成功制备EBOV-GP蛋白晶体,并借助X射线衍射实验得到其三维结构(PDB ID为5KEN),提供了一种计算机高通量虚拟筛选模型,可用于鉴定靶向潜在抑制剂。为了缩短药物前期研发周期,本研究采用计算机模拟技术,在选定虚拟筛选靶点前提下,采用类药性评估、分子对接以及吸收、分布、代谢和排泄(absorption、distribution、metabolism and excretion,ADME)预测等多尺度虚拟筛选策略,然后运用进行分子动力学模拟,从中国台湾中医药数据库TCM@TAIWAN中筛选潜在的先导化合物,并分析候选分子与靶点的相互作用情况。
1 方法
1.1 资料与计算方法
分子建模、能量优化、类药性参数计算来源CambridgeSoft公司ChemBioOffice 2016;分子对接计算均采用BIOVIA公司开发的Discovery Studio 2016的Libdock模块与CDOCKER模块、剑桥晶体学数据中心CCDC的GOLD Suite 5.3软件包;ADME参数计算采用Schrodinger 2018的QikProp 模块;分子动力学模拟及MM/PBSA结合自由能预测均采用GROMACS 2019程序包。
1.2 靶蛋白EBOV-GP的预处理与活性口袋的确定
EBOV-GP介导病毒进入宿主细胞,病毒内在化为内体后,GP被宿主半胱氨酸蛋白酶裂解,暴露出受体结合位点(receptor binding site,RBS),被机体的免疫监视[3]。Onawole等[4]利用AutoDock Vina与FlexX软件包对MCULE配体库(包含36 000 000个小分子)进行高通量虚拟筛选,并结合类药性及毒理学预测,得到3个候选活性化合物,命名为Hit-SC1、Hit-SC2、Hit-SC3。比较3个阳性抑制剂与EBOV-GP的结合模式,发现候选化合物与EBOV-GP活性位点的主要作用力是氢键与疏水相互作用,并且在氢键发生作用的关键氨基酸残基为Glu578、Ser583、Asn586、Thr577和Arg580。如图1所示,所有残基位于靶蛋白跨膜区外侧,均围绕在病毒糖蛋白三聚体围绕形成的口袋,一旦有小分子抑制剂进入活性口袋区域,可以有效阻止外来病毒粒子附着并侵入宿主细胞。位于活性口袋的9个氨基酸残基Arg587、Ile590、Leu579、Phe582、Asp127、Leu165、Ile129、Asn98和Thr576与3个候选活性化合物发生弱的疏水相互作用或离子键。
A-活性口袋示意图 B-活性部位的关键氨基酸示意图
以“Ebola virus”“EBOV-GP”为关键词,检索蛋白质结构数据库PDB,查询EBOV-GP晶体结构,确定适合做虚拟研究的蛋白结构有4个,依次为6NAE、6HRO、5KEL、5KEN。6NAE与6HRO是跨膜糖蛋白单体结构,5KEL与5KEN是跨膜糖蛋白三聚体结构[2]。目前糖蛋白单体结构是埃博拉病毒虚拟筛选的重要结构靶点,但是糖蛋白三聚体结构为目前处于临床试验中的潜在疫苗的主要靶点,已有[5]、[6]、[7]、[8]等多家顶级权威国际杂志发文证实其靶点的重要性,因此本研究选择埃博拉病毒跨膜糖蛋白三聚体结构形成活性空腔作为虚拟筛选的研究靶点,筛选合适的药效分子。
基于以上分析,本研究从PDB蛋白质结构数据库下载蛋白晶体结构(PDB ID为5KEN),去除结构中附着的中和抗体C2G4(临床候选免疫治疗剂);使用Schrodinger 2018软件的Protein Preparation wizard模块[9]对5KEN结构进行修正,在OPLS3力场下对蛋白进行能量优化,晶体结构重原子RMSD收敛标准为0.05 nm;使用Receptor Grid Generation模块[10]计算包含结合中心和尺寸的网络系统,得出结合中心点、、轴的坐标分别为159.734 8、159.927 1、156.096 8,结合空间维数均为20,所使用的结合参数确保整个靶蛋白活性口袋被封闭在网格中;选择Asn98、Glu578、Phe582、Ser583、Asn586作为关键氨基酸残基,并关注Leu165、Thr576、Thr577、LeuB579、Arg580、Ile590与候选药效分子是否产生疏水相互作用、离子键等。
1.3 中药分子化学结构的预处理
TCM@TAIWAN数据库[11]是目前全世界最大的提供下载虚拟筛选的中医药小分子数据库,由中国台湾中国医药大学计算与系统生物学实验室创立,涵盖443种中药材的化学成分以及二维、三维结构数据。本研究采用TCM@TAIWAN中36 064个化合物作为筛选的配体库,采用Schrodinger 2018的Ligprep模块对各化合物分子进行加氢、能量最小化、几何优化等处理;在力场OPLS3[12]下,计算pH值为(7.0±2.0)的化合物离子状态,搜索合理构象作为对接起始构象,形成可能的互变异构体,并生成批量预处理文件(*.sdf);Ahmad等[13]使用同源建模EBOV-GP的蛋白质空间结构,进行分子对接、分子动力学模拟等计算机模拟实验,最终找到候选药效分子胺碘酮,因此本研究将胺碘酮作为阳性对照药物。
1.4 基于类药性、分子对接、ADME与结合自由能预测的多尺度虚拟筛选
1.4.1 基于类药性规则“Lipinski Ro5[14]”“Verber Ro3[15]”的第1轮筛选 对中药分子配体库进行筛选,挑选出19 981个符合Lipinski五原则的分子,具备良好的药动学参数、潜在的成药性、较高的口服生物利用度等特质。
1.4.2 基于高通量筛选程序LibDock[16]的第2轮筛选 根据小分子构象与受体相互作用热区匹配的原理,LibDock将小分子构象刚性对接到受体的结合口袋中,适合于对大规模数据库进行快速精确的虚拟筛选。将EBOV-GP与第1轮筛选得到的19 981个分子逐一进行分子对接,参照打分函数对所有分子进行排序,并以胺碘酮对接打分值作为阈值,最终选择12 982个化合物,筛选得到候选分子的LibDock-Score均高于116.77。
1.4.3 基于半柔性对接程序GOLD[17]的第3轮筛选 GOLD源于剑桥晶体学数据中心,具有高度可配置性,可充分利用蛋白质-配体系统的背景知识,最大限度地提高对接性能。精确度参数设置Very Flexible,评分函数设置GoldScore,其他参数选择默认值;以胺碘酮对接打分值(−69.79 kJ/mol)作为阈值,最终选择672个化合物,筛选得到候选分子的GoldDock-Score均低于−69.79 kJ/mol。
1.4.4 基于高温动力学柔性对接程序CDOCKER[18]的第4轮筛选 基于分子力场CHARMm[19]的对接程序,采用soft-core potentials以及optional grid representation将配体分子与受体活性位点进行对接。首先采用高温动力学的方法随机搜索小分子构象,随后采用模拟退火的方法将各个构象在受体活性位点区域进行优化,从而产生高精度的对接结果。对672个候选分子的CDOCKER_ENERGY进行排序,以胺碘酮的对接打分值(−211.25 kJ/moL)作为参考值,选取打分排序前96的分子作为候选分子,其CDOCKER_ENERGY均高于202.13 kJ/moL,与阳性对照药物的参数基本一致。
1.4.5 基于ADME筛选 采用Schrodinger的QikProp模块基于ADME药动学参数对96个候选分子进行筛选,除去吸收代谢差的分子。从相对分子质量、氢键供体、氢键配体、溶剂可及表面积、HERG_IC50、水溶性、脂水分配系数、口服生物利用度等多个方面预测评估,共筛选得到32个候选化合物。
1.4.6 基于MM/GBSA结合自由能筛选 进行MM/GBSA[20]算法优化排序,进一步提高候选分子与受体蛋白结合自由能的计算精度。以胺碘酮与受体蛋白的结合能(−333.17 kJ/mol)作为阈值,最终选择8个候选分子。
1.5 分子动力学模拟
为了从动力学和热力学角度分析EBOV-GP蛋白受体与候选抑制剂分子的相互结合模式,以及靶点-配体对接后构象的其他信息随着时间变化的具体情况,使用GROMACS 2019[21]进行分子动力学模拟。EBOV-GP蛋白的拓扑参数取自Amber的FF03[22]力场,由PDB2GMX工具获得;小分子抑制剂各原子的部分电荷先用Gaussian 09[23]在HF/6-31G*水平计算静电势,再用Amber中的RESP[24]电荷拟合程序算出;于26.85 ℃对蛋白和小分子配体复合物进行1000步的能量最小化,对优化后的体系分别进行100 ps的NVT和NPT系综平衡,限制体系位置;系统温度保持26.85 ℃,进行30 ns的动力学模拟,时间间隔为2 fs,每隔10 ps保存1次坐标轨迹文件;从15~30 ns稳定的动力学轨迹中提取150帧动力学轨迹文件,使用MM/PBSA[25]计算体系稳定后的结合自由能。
2 结果
2.1 虚拟筛选结果分析
基于靶点EBOV-GP的三维空间结构,对TCM@TAIWAN数据库中的36 064个天然化合物进行多尺度虚拟筛选,最终确定打分排名前4的化合物ZINC85531496、ZINC85567560、ZINC85592968、ZINC33833122作为候选单体,见表1、2。
表1 排名前4的候选化合物与靶点的分子对接打分值与MM/GBSA结合能
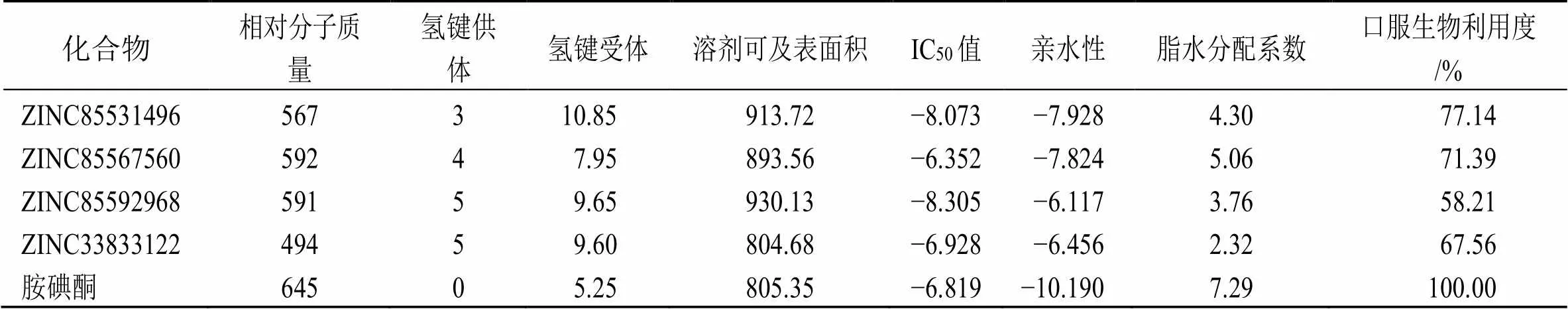
表2 排名前4候选化合物的ADME参数预测值
2.2 候选化合物与靶点的相互作用位点分析
采用Discovery Studio Visualizer软件绘制以氢键、疏水相互作用、离子相互作用和水桥等蛋白质-配体不同形式相互作用的分子对接结果。如图2所示,阳性对照药物胺碘酮与EBOV-GP之间的主要作用力是常规氢键、盐桥、alkyl相互作用;与EBOV-GP F链氨基酸残基Arg587、M链氨基酸残基Asn586均形成氢键,并且均为常规氢键类型;与EBOV-GP B链Glu578、F链Glu578、M链Glu578均形成盐桥相互作用;与EBOV-GP空腔的碱性氨基酸残基M链Arg587、M链Arg590形成alkyl相互作用;与EBOV-GP F链碱性氨基酸Arg587形成π-alkyl堆积作用。
如图3所示,ZINC85531496与EBOV-GP之间的主要作用力是氢键、π-cation相互作用、π-anion相互作用、π-π堆积作用;与EBOV-GP B链、F链、M链氨基酸残基Ser583、Glu578均形成氢键,与B链形成2个常规氢键,与F链、M链均形成2个碳氢键;与EBOV-GP F链Glu578、M链Glu578均形成π-anion相互作用;与EBOV-GP F链Arg580形成π-cation相互作用,与F链Phe582形成2个π-π堆积作用。
如图4所示,ZINC85567560与EBOV-GP之间的相互作用不仅有氢键、范德华力,还有π-π堆积作用、π-anion相互作用、π-alkyl相互作用。ZINC85567560与EBOV-GP氨基酸残基M链Ser583、M链和F链Glu578形成常规氢键,与B链Ser583形成双碳氢键,与B链Phe582形成π-π堆积作用,与B链Glu578形成π-anion相互作用,与F链Phe582形成π-alkyl相互作用。
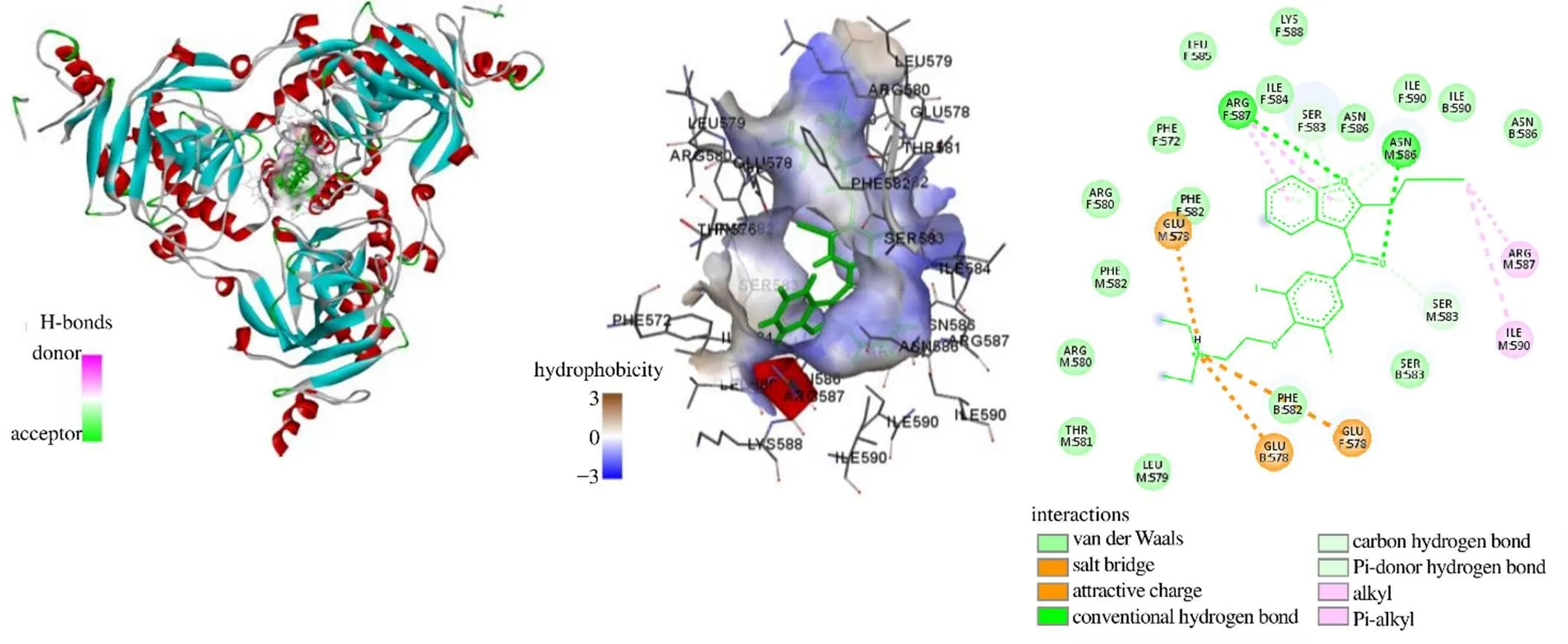
图2 EBOV-GP与胺碘酮的分子对接
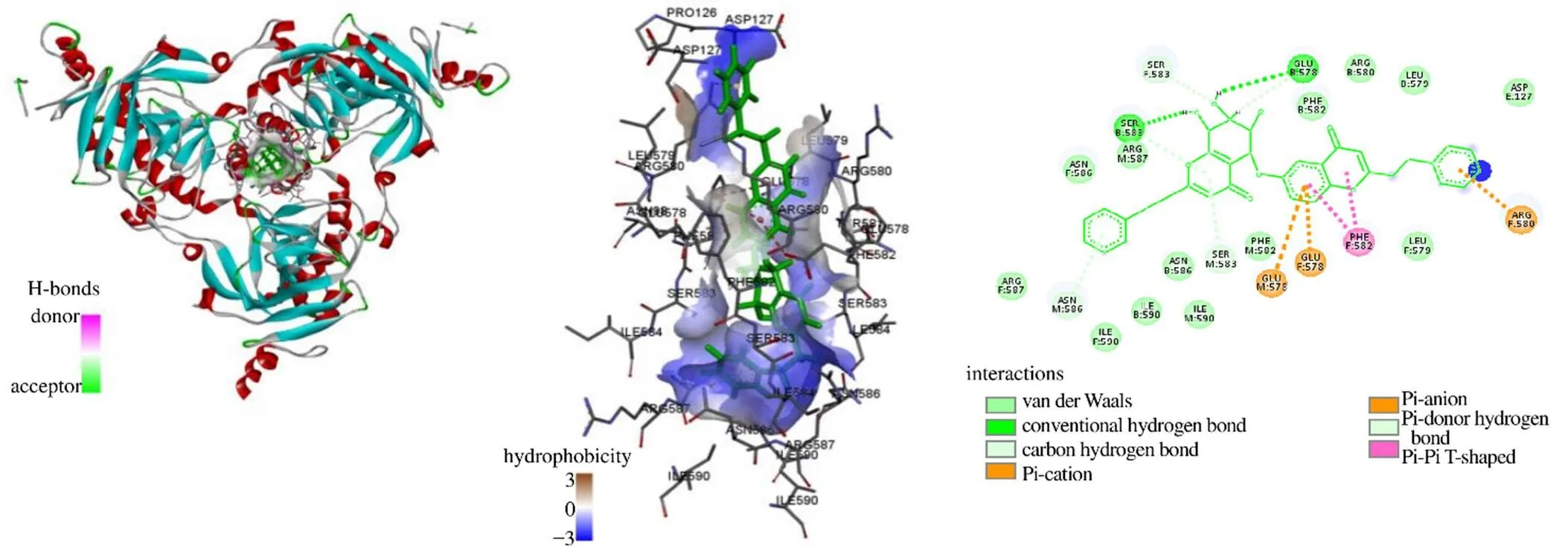
图3 EBOV-GP与ZINC85531496的分子对接
如图5所示,ZINC85592968与EBOV-GP之间的相互作用不仅有氢键,还有π-anion相互作用与π-alkyl相互作用。ZINC85592968与EBOV-GP M链Thr577形成常规氢键,与M链Glu578、B链Ser583、M链Ser583、B链Phe582、F链Glu578、F链Ser583均形成1个碳氢键,与B链Glu578形成π-anion相互作用,与M链Arg580、Leu579分别形成π-alkyl相互作用。
如图6所示,ZINC33833122与EBOV-GP之间的相互作用不仅有氢键,还有π-anion相互作用与π-alkyl相互作用。ZINC33833122与EBOV-GP B链Thr577形成常规氢键,与M链Glu578、B链Ser583、M链Ser583、B链Phe582、F链Glu578、F链Ser583均形成1个碳氢键,与B链Glu578形成π-anion相互作用,M链Arg580、Leu579分别形成π-alkyl相互作用。这些作用力使结合能降低,亲和力增加,候选单体与EBOV-GP三聚体空腔部位紧密结合,形成稳定的复合物,可以有效阻止外来病毒粒子附着并侵入宿主细胞。
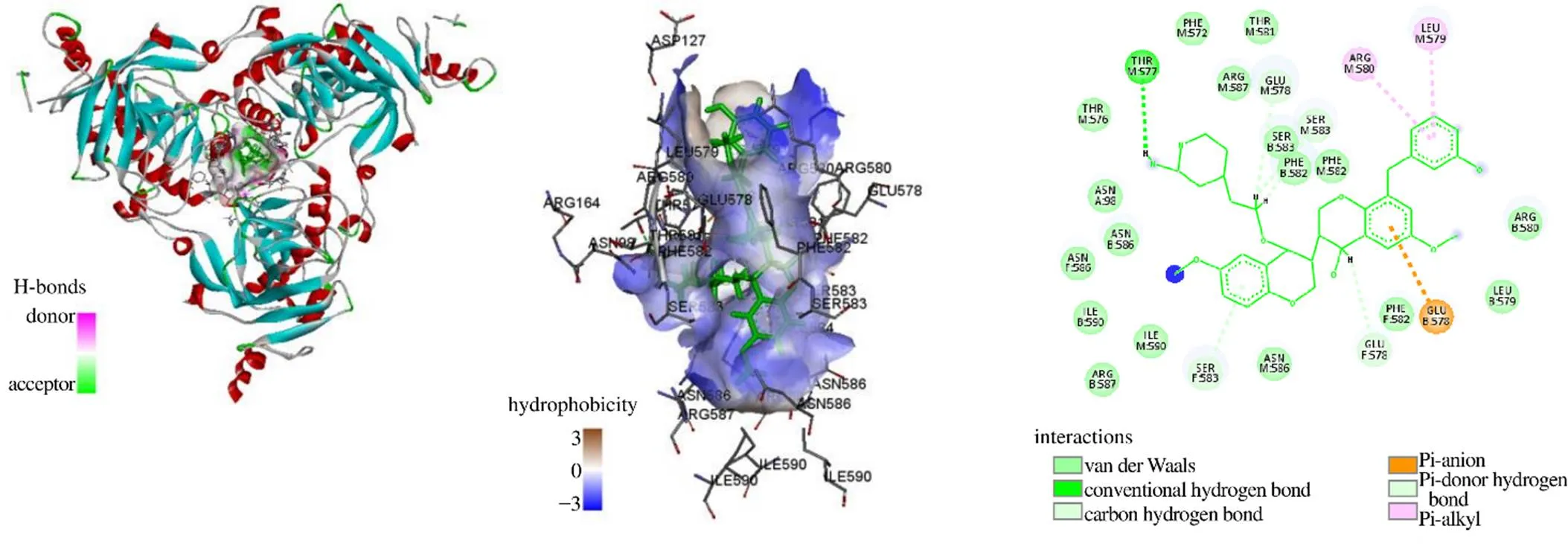
图5 EBOV-GP与ZINC85592968的分子对接
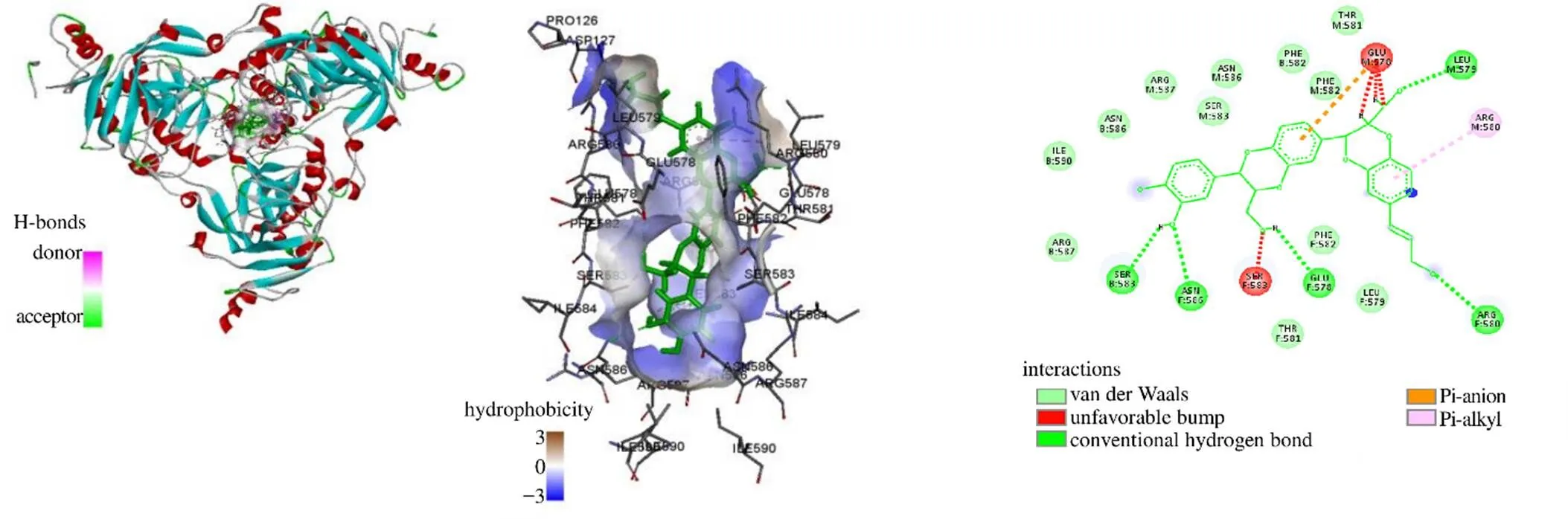
图6 EBOV-GP与ZINC33833122的分子对接
氢键在生物大分子中起着重要作用,氢键是蛋白质与配体相互作用的主要推动力之一,在稳定蛋白质-配体复合物方面起着不可或缺的作用[26]。胺碘酮与EBOV-GP氨基酸残基Arg587、Asn586形成氢键相互作用,产生的氢键数目为2个;4个候选单体均与EBOV-GP氨基酸残基Glu578、Ser583形成氢键相互作用,甚至形成双氢键,这些残基均是候选单体的氢键受体,并且常规氢键与碳氢氢键总数均≥5(其中ZINC85531496、ZINC85567560、ZINC85592968、ZINC33833122氢键总数分别为6、5、6、5个)。胺碘酮与EBOV-GP氨基酸残基Glu578形成盐桥相互作用,与碱性氨基酸残基Arg587、Arg590形成alkyl相互作用,与碱性氨基酸Arg587形成π-alkyl堆积作用。4个候选单体均与EBOV-GP氨基酸残基Glu578形成π-anion相互作用,与碱性氨基酸Arg580或Leu579形成π-alkyl相互作用,这些相互作用在候选配体分子与EBOV-GP三聚体围绕形成的结合腔的残基结合中起到辅助作用。总体而言,与阳性对照药物胺碘酮相比,4个候选分子与EBOV-GP的结合更加具有优势,均具有更强的相互作用,推测可能具有更高的抑制作用。
2.3 分子动力学模拟结果
如图7所示,15 ns后所有体系的均方根偏差(root-mean-square deviation,RMSD)波动值均稳定在0.1 nm以下,模拟的分子动力学模拟体系均达到平衡状态,表明模拟结果可用于进一步分析。
通过计算化合物与受体蛋白复合物氨基酸骨架原子在分子动力学模拟过程中的均方根波动值(root mean square fluctuation,RMSF),分析EBOV-GP与4种候选化合物骨架氨基酸在模拟过程中的波动性。如图8所示,ZINC85531496-GP、ZINC85567560-GP、ZINC85592968-GP、ZINC33833122- GP氨基酸波动变化趋势与蛋白质受体APO大致相似,在氨基酸555~595区间出现较低的波动性(RMSF<0.15 nm),且ZINC85531496-GP与ZINC33833122-GP的波动性明显低于蛋白质受体APO;ZINC85531496-GP、ZINC85567560-GP、ZINC85592968-GP在氨基酸520~530、540~550区间出现大幅波动(RMSF>0.15 nm),其中ZINC85531496-GP、ZINC85592968-GP波动幅度明显高于蛋白质受体APO,ZINC85567560-GP与蛋白质受体APO具有相似的波动轨迹;ZINC33833122- GP的氨基酸波动变化趋势不同其他3个体系,氨基酸538~545区间出现极高的波动峰值(RMSF>0.3 nm)。以上4个配体-靶蛋白复合物体系中RMSF波动较小的氨基酸与关键氨基酸相吻合,可能与化合物和EBOV-GP之间产生了较稳定的氢键、疏水作用或π-π堆积作用有关,可以将化合物与结合口袋周围的关键氨基酸残基侧链相互连接在一起,形成较稳定的三聚体复合物;波动幅度较大的区域位于蛋白单体与单体结合的铰链区,增加了抗原表位的异质性,更加易于和抗体蛋白结合。
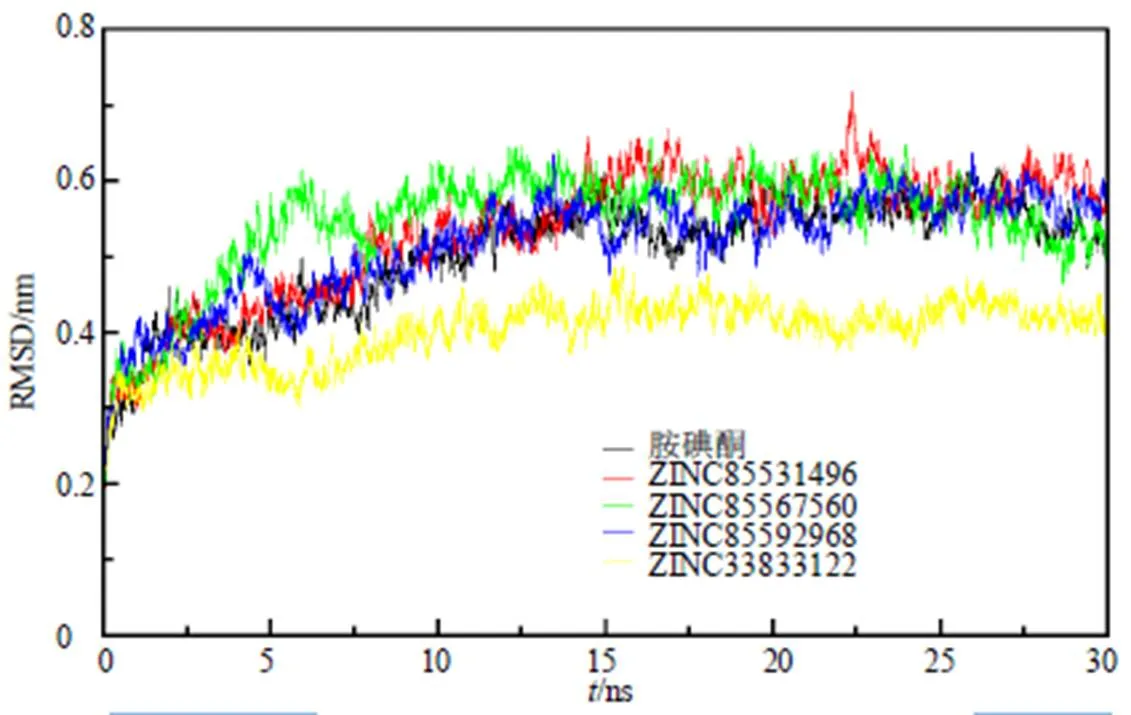
图7 EBOV-GP的氨基酸骨架原子随时间变化的RMSD值
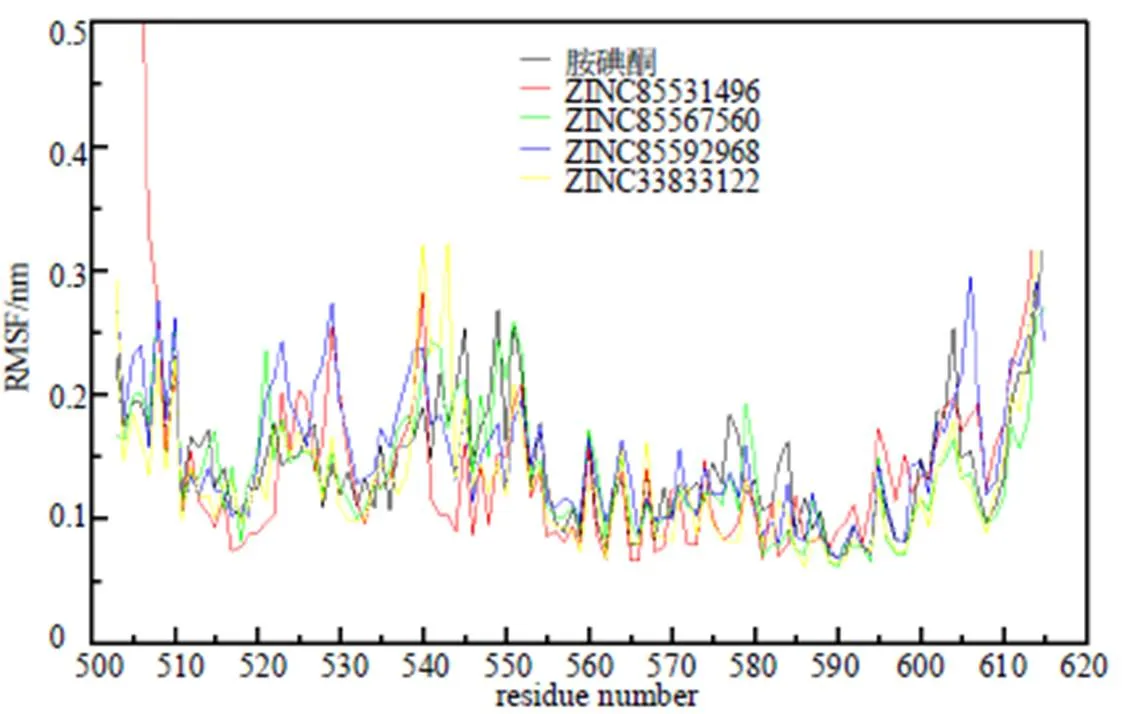
图8 配体-蛋白复合物氨基酸骨架原子的RMSF值波动图
进一步研究ZINC85531496、ZINC85567560、ZINC85592968、ZINC33833122与EBOV-GP之间的结合亲和力,采用MM/PBSA方法计算4种化合物与EBOV-GP复合物的结合自由能,分析4种化合物与EBOV-GP结合的作用。如表3所示,范德华力与静电势能最有利于4种化合物与EBOV-GP结合,表面溶剂化作用也有利于结合,但与范德华力和静电势能的数值相比作用较弱;极性溶剂化作用不利于体系结合。ZINC85592968与ZINC85567560具有较强的范德华力,但由于ZINC85592968具有最弱的静电势能,导致其最终的结合自由能不占优势,排名第3;ZINC85531496与ZINC33833122具有近似的范德华力数值,但ZINC33833122具有较高的极性溶剂化作用,非常不利于配体与受体结合,因此与蛋白质结合过程中由于溶剂中的极性溶剂化相互作用很大程度上抵消了静电势能与范德华力之和,导致其最终的结合自由能排名最低;ZINC85531496与ZINC85567560的非极性相互作用更有利于配体和受体的结合,其中范德华力与静电势能均起主导作用,均有利于配体与受体的结合,并且极性溶剂化作用产生的负作用较弱,这也是ZINC85531496、ZINC85567560与EBOV-GP结合自由能具有优势效应的关键因素。
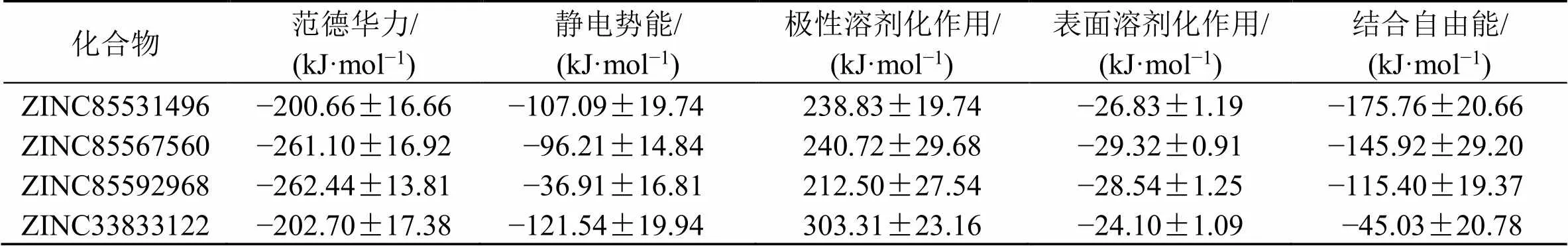
表3 4种候选化合物与EBOV-GP的MM/PBSA结合自由能
结合自由能数值越小,表示结合所需能量越低,越有利于配体与蛋白的结合。通过比较4种化合物与EBOV-GP的结合自由能,得出蛋白配体结合强弱关系为ZINC85531496-GP>ZINC85567560-GP>ZINC85592968-GP>ZINC33833122-GP,与分子对接结果一致。
3 讨论
EBOV最初在西非局部地区反复爆发,2014年蔓延至整个西非,造成超过11 000人死亡。刚果民主共和国作为第2大流行国家,已报道2264人死亡,EBOV为国际关注的突发公共卫生事件[27]。针对EBOV的疫苗或药物的研发仍处于动物模型实验或人体临床初试阶段。临床试验中的候选免疫治疗剂ZMapp™,包含2个EBOV-GP的特异性抗体(c2G4、c4G7)和1个GP/SGP-交叉反应抗体(c13C6),ZMapp™的制备既昂贵又费时,并且其抗体表位之间的差异以及有效免疫的详细机制仍然难以确定,尚未经临床试验验证其有效性[10]。目前EBOV相关疫苗和药物的研发围绕跨膜糖蛋白三聚体GP结构,主要包括GP1(受体结合)和GP2(病毒融合)2个亚基,EBOV-GP的C末端区域包含381个氨基酸,可以组装成高度糖基化的黏蛋白样结构域(mucin-like domain,MLD),介导与宿主细胞的结合并进入宿主细胞,因此,EBOV-GP为重要的药物靶点[28]。
EBOV-GP是丝状病毒科I类融合蛋白,由二硫键互连的GP1与GP2 2个亚基组成,在病毒体表面上形成同型三聚体活性分子。GP1由核心颗粒、聚糖帽和MLD 3部分组成,GP2包含膜融合结构域,并锚定在膜上。EBOV-GP是一种动态元件,从融合前的亚稳态构象不可逆转地折叠成融合后的稳态构象,从而驱动病毒与宿主细胞融合。融合过程需要5个重要条件:①与细胞表面受体(如TIM-1、Axl、硫酸乙酰肝素及DC-SIGN)结合;②胞饮作用;③组织蛋白酶在低pH下裂解GP;④结合内体中NPC1胆固醇转运蛋白;⑤驱动GP发生膜融合所需的构象变化。其中驱动GP发生膜融合所需的构象变化是最有可能的最终触发关键因素,因此有效抑制EBOV-GP构象转变可以阻断病毒与宿主细胞融合。
本研究基于EBOV-GP的三维空间结构,运用计算机虚拟筛选技术从TCM@TAIWAN数据库中寻找抗病毒的中药活性分子。Ahmad等[13]仅采用Auto Dock Vina与Flex-X 2种分子对接软件从Mcule化合物库(https://mcule.com/database/)进行虚拟筛选,ADME药动学评价仅使用ADMET-SAR进行过滤。本研究在此基础上,重新设计方法,优化虚拟筛选的精度。
首轮筛选遵循类药性指标Lipinski Ro5、Verber Ro3作为通过筛选大型化学结构文库表征命中规则的重要标准,违反1种以上类药性参数的化合物可能存在生物利用度问题,因此16 083种化合物被剔除,挑选出19 981种化合物。随后第2轮筛选采用业界公认的3种分子对接软件LibDock、GOLD、CDOCKER进行低-中-高3种精度的虚拟筛选,逐步缩小候选化合物库的规模,极大地提高了计算精度,尽可能降低虚拟筛选假阳性率。随后第3轮筛选采用Schrodinger软件的QikProp模块来完成ADME参数预测,较ADMET-SAR进行过滤策略,筛选规则新增了肝毒性、口服生物利用度2项关键ADMET指标,并考虑了Lipinski Ro5、Verber Ro3 2项类药性指标,使得本研究的ADME评价体系更加客观、系统、精准。为了模拟0.9%氯化钠溶液中靶点蛋白与命中分子的结合模式,本研究采用高性能GPU加速技术进行了30 ns时长的分子动力学模拟,并使用MM/GBSA和MM/PBSA 2种算法计算命中分子与靶点的结合自由能,与常规的分子对接后结合能结果相比较,富集精准度被大幅提升。本研究尚处于药物开发的前期阶段,缺乏抗病毒活性验证、药动学评估等数据支撑,后期将联合专业病毒学研究平台,开展相关生物学活性验证及安全性评价。
综上,本研究创新地采用多种计算机虚拟筛选技术挖掘中药来源的抑制剂分子,ZINC85531496、ZINC85567560、ZINC85592968、ZINC33833122能够有效结合EBOV-GP,抑制其构象转变,从而阻断病毒与宿主细胞膜融合,为从天然产物化学结构数据库中挖掘阻断埃博拉病毒侵染人体的先导化合物提供一定参考。
利益冲突 所有作者均声明不存在利益冲突
[1] Rojas M, Monsalve D M, Pacheco Y,. Ebola virus disease: An emerging and re-emerging viral threat [J]., 2020, 106: 102375.
[2] Pallesen J, Murin C D, de Val N,. Structures of Ebola virus GP and sGP in complex with therapeutic antibodies [J]., 2016, 1(9): 16128.
[3] Bornholdt Z A, Ndungo E, Fusco M L,. Host-primed Ebola virus GP exposes a hydrophobic NPC1receptor- binding pocket, revealing a target for broadly neutralizing antibodies [J]., 2016, 7(1): e02154-e02115.
[4] Onawole A T, Kolapo T U, Sulaiman K O,. Structure based virtual screening of the Ebola virus trimeric glycoprotein using consensus scoring [J]., 2018, 72: 170-180.
[5] Stanley D A, Honko A N, Asiedu C,. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against Ebolavirus challenge [J]., 2014, 20(10): 1126-1129.
[6] Henao-Restrepo A M, Longini I M, Egger M,. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: Interim results from the Guinea ring vaccination cluster-randomised trial [J]., 2015, 386(9996): 857-866.
[7] Tapia M D, Sow S O, Lyke K E,. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: A phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial [J]., 2016, 16(1): 31-42.
[8] Ledgerwood J E, DeZure A D, Stanley D A,. Chimpanzee adenovirus vector Ebola vaccine [J]., 2017, 376(10): 928-938.
[9] Sastry G M, Adzhigirey M, Day T,. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments [J]., 2013, 27(3): 221-234.
[10] Friesner R A, Murphy R B, Repasky M P,. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes [J]., 2006, 49(21): 6177-6196.
[11] Chen C Y. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico [J]., 2011, 6(1): e15939.
[12] Harder E, Damm W, Maple J,. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins [J]., 2016, 12(1): 281-296.
[13] Ahmad N, Farman A, Badshah S L,. Molecular modeling, simulation and docking study of Ebola virus glycoprotein [J]., 2017, 72: 266-271.
[14] Lipinski C A, Lombardo F, Dominy B W,. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings [J]., 2001, 46(1/2/3): 3-26.
[15] Veber D F, Johnson S R, Cheng H Y,. Molecular properties that influence the oral bioavailability of drug candidates [J]., 2002, 45(12): 2615-2623.
[16] Rao S N, Head M S, Kulkarni A,. Validation studies of the site-directed docking program LibDock [J]., 2007, 47(6): 2159-2171.
[17] Verdonk M L, Cole J C, Hartshorn M J,. Improved protein-ligand docking using GOLD [J]., 2003, 52(4): 609-623.
[18] Gagnon J K, Law S M, Brooks C L 3rd. Flexible CDOCKER: Development and application of a pseudo-explicit structure-based docking method within CHARMM [J]., 2016, 37(8): 753-762.
[19] Vanommeslaeghe K, Hatcher E, Acharya C,. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields [J]., 2010, 31(4): 671-690.
[20] Zhang X H, Perez-Sanchez H, Lightstone F C. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin [J]., 2017, 17(14): 1631-1639.
[21] Abraham M J, Murtola T, Schulz R,. GROMACS: High performance molecular simulations through multi- level parallelism from laptops to supercomputers [J]., 2015, 1/2: 19-25.
[22] Khoury G A, Bhatia N, Floudas C A. Hydration free energies calculated using the AMBER ff03 charge model for natural and unnatural amino acids and multiple water models [J]., 2014, 71: 745-752.
[23] 黎唯, 谢惠定, 黄燕, 等. Gaussian09/GaussView5.0在分析化学教学中的应用 [J]. 昆明医科大学学报, 2016, 37(10): 134-136.
[24] Cieplak P, Cornell W D, Bayly C,. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins [J]., 1995, 16(11): 1357-1377.
[25] Kongsted J, Ryde U. An improved method to predict the entropy term with the MM/PBSA approach [J]., 2009, 23(2): 63-71.
[26] Du X, Li Y, Xia Y L,. Insights into protein-ligand interactions: Mechanisms, models, and methods [J]., 2016, 17(2): E144.
[27] Rugarabamu S, Mboera L, Rweyemamu M,. Forty-two years of responding to Ebola virus outbreaks in Sub-Saharan Africa: A review [J]., 2020, 5(3): e001955.
[28] Kiiza P, Mullin S, Teo K,. Treatment of Ebola-related critical illness [J]., 2020, 46(2): 285-297.
Mining antiviral active inhibitors in traditional Chinese medicine targeted with transmembrane glycoprotein of Ebola virus bydiscovery
SHI Hai-long, SHI Yong-heng, WANG Chuan, CHENG Yi, HUANG Yue, LIU Ji-ping, WANG Bin
Shaanxi University of Chinese Medicine, Xianyang 712046, China
Using computer virtual screening technology, some inhibitors of transmembrane glycoprotein of Ebola virus (EBOV-GP) would be sorted from TCM@TAIWAN Database.Taking EBOV-GP as the target, using multi-scale virtual screening strategies such as drug-like evaluation, molecular docking, ADME prediction, molecular dynamics simulation, etc., the candidates were mined from TCM@TAIWAN database, and then the interaction between candidates and targets was analyzed.Amiodarone was used as a positive control, and four monomers (ZINC85531496, ZINC85567560, ZINC85592968 and ZINC33833122) from Chinese medicine with good drug-like properties were selected, and their affinity and interaction with EBOV-GP were superior to amiodarone.A variety of computer virtual screening technologies to mine EBOV-GP inhib are used to facilitate the extraction, design, and experimental synthesis of anti-Ebola virus inhibitors from natural products chemical structure database.
Ebola virus; transmembrane glycoprotein; virtual screening; Chinese medicine; antiviral active molecule
R285.51
A
0253 - 2670(2021)13 - 3933 - 10
10.7501/j.issn.0253-2670.2021.13.017
2020-11-30
陕西省中医管理局中医药科研课题(JCMS003);陕西省自然科学基础研究计划项目(2019JQ-401);陕西中医药大学创新团队项目(2019-YL13);国家级大学生创新创业项目(201910716031)
史海龙(1981—),男,博士,美国UNMC博士后,研究方向为计算机辅助药物设计及生物大分子模拟。Tel: 15829489958 E-mail: shl112@sntcm.edu.cn
王 斌,教授,硕士生导师,主要从事防治心脑血管疾病的方药研究。E-mail: wangbing812@126.com
[责任编辑 李亚楠]
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
材料科学与工程学报(2016年4期)2017-01-15
中学化学(2015年12期)2016-01-19
合成化学(2015年4期)2016-01-17
池州学院学报(2015年3期)2016-01-05
天津科技大学学报(2015年2期)2015-08-09
原子与分子物理学报(2014年3期)2014-02-28
无机化学学报(2014年6期)2014-02-28
无机化学学报(2014年5期)2014-02-28