射干叶绿体基因组结构、序列特征与系统发育分析
2021-07-15 11:26王军峰黄雯馨应梦豪马佳莹戴晨宇
中草药 2021年13期
蒋 明,王军峰,朱 晏,黄雯馨,应梦豪,马佳莹,戴晨宇
射干叶绿体基因组结构、序列特征与系统发育分析
蒋 明1,王军峰2,朱 晏1,黄雯馨1,应梦豪1,马佳莹1,戴晨宇1
1. 台州学院生命科学学院,浙江 台州 318000 2. 华东药用植物园科研管理中心,浙江 丽水 323000
以射干为材料,在测序、组装获得叶绿体基因组的基础上,明确其结构、序列特征及系统发育关系。利用PE150双末端策略进行建库测序,用NOVOPlasty组装完整的叶绿体基因组,经PCR验证边界,借助生物信息学工具进行序列分析和系统发育研究。射干的叶绿体基因组全长为153 816 bp,大单拷贝区、反向重复区和小单拷贝区的长度分别为83 143、26 214、18 245 bp。射干叶绿体基因组共有133个基因,编码基因、tRNA和rRNA的数量分别为92、38和8;有2个拷贝,其中一个为假基因。系统发育分析结果表明,7种植物叶绿体基因组在发育树上可分为4组,射干与同为鸢尾科的溪荪聚为一组,支持率达100%。射干叶绿体基因组的组装、序列分析和系统发育分析,为该药用植物的遗传结构和遗传多样性研究奠定了基础。
射干;叶绿体基因组;结构;序列分析;鸢尾科
射干(L.) Redouté为鸢尾科(Iridaceae)射干属多年生草本植物,具结节状的根状茎,叶片剑形,花橙红色,花瓣上散生紫褐色斑点,分布于我国的吉林、山东、安徽、江苏、浙江和福建等20多个省份[1]。射干分布广泛,是一种十分常见的药用植物,有着悠久的栽培和药用历史;射干的主要药用部位为其根状茎,它富含酚类化合物,尤其是类黄酮和异黄酮类物质,如射干苷、白射干素、鸢尾黄素和野鸢尾苷等[2]。射干具有清热解毒、散结消炎和利咽消肿等功效,可用于治疗扁桃腺炎、喉痹咽痛和腰痛等[3-5]。射干叶形优美、花色亮丽、花期较长,具有较好的观赏价值,常用于盆栽、庭院美化或药草园片植等,是一种观赏和药用兼用植物[6]。近年来,有关射干的研究主要集中在栽培技术、组培快繁、病虫害防治、化学成分、药理作用和转录组等方面[7-11]。
叶绿体是绿色植物进行光合作用和能量转换的细胞器,它将太阳能转换为化学能,用于物质积累和植物的生长发育[12]。叶绿体在植物逆境防御中也起着十分重要的作用,在不良环境下,产生大量的活性氧(reactive oxygen species,ROS),ROS作为一种调控基因表达水平的信号分子,促使植物产生新的适应[13-14]。叶绿体基因组以双链环状形式存在于叶绿体中,被子植物的叶绿体基因组大小为120~170 kb,测序和组装比核基因组容易[15]。随着测序技术的快速发展及测序费用的降低,越来越多的叶绿体基因组得以注释,目前已有1000多种植物完成叶绿体基因组的测序,它们在遗传多样性、基因组进化、基因水平转移、系统发育分析和群体遗传学等方面得到了应用[16-18]。目前,有关射干叶绿体基因组的测序、组装和注释等研究未见报道。本研究以射干叶片DNA为材料,在利用高通量测序的基础上拼接叶绿体基因组,明确其序列特征、基因组结构及与近缘特种间的系统发育关系,为后续开展遗传多样性和群体遗传学研究奠定基础。
1 材料与仪器
1.1 材料
射干叶片采自浙江舟山东福山岛,伴生植物有山菅(L.) DC.、小茄Thunb.、藓状景天Hemsl.和滨海前胡Thunb.等。采集健康叶片,装入样品袋后带回实验室备用。
1.2 仪器
艾本德Eppendorf移液枪;超净工作台(苏州安泰空气技术有限公司);ThinkPad P52移动工作站;伯乐C1000型PCR仪(Bio-Rad公司,美国);Covaris超声波DNA破碎仪(Chromatin Shearing,美国);Illumina HiSeq X Ten测序仪;北京六一DYY-12型电泳仪及电泳槽(北京市六一仪器厂);伯乐Gel Doc XR+凝胶成像系统(Bio-Rad公司,美国)。
2 方法
2.1 DNA的提取和文库构建
在无菌研砵中加入适量液氮,用研棒将叶片磨成细粉末,再利用十六烷基三甲基溴化铵(Cetyltrimethylammonium bromide,CTAB)方法提取基因组DNA,经电泳检测后用于构建文库。用超声波破碎仪将基因组DNA片段化,经末端修复、添加A尾、两端加测序接头、产物纯化及PCR扩增等过程,完成文库的构建。
2.2 高通量测序
采用双端(paired-end,PE)策略,用Illumina HiSeq X Ten高通量测序仪进行测序,读长为2×150 bp,共获得3.55 G原始数据。利用NGS QC Toolkit v2.3.3对原始数据进行过滤,去除接头和低质量的序列[19]。最终共得到11 814 172条clean reads,20值达97.53%,30为92.73%,序列质量较高,可用于后续的拼接和注释。
2.3 叶绿体基因组的拼接和注释
叶绿体基因组的拼接在ThinkPad P52移动工作站上进行,拼接采用NOVOPlasty程序[20]。利用在线工具DOGMA(dual organellar genoMe annotator)对序列进行基因注释,网址为http:// dogma.ccbb.utexas.edu/,起始和终止密码子通过手工方式进行调整[21]。tRNA用tRNAscan-SE(http:// www.lowelab.ucsc.edu/tRNAscan-SE/)和ARAGORN预测[22-23]。OGDRAW(organellar genome draw)用于生成叶绿体基因组圈图,网址为http://www. ogdraw. mpimp-golm.mpg.de[24]。
2.4 边界序列的PCR克隆和测序验证
叶绿体基因组的4个边界采用PCR方法进行鉴定,根据拼接的草图,共设计了4对引物,分别是YGUP1:5’-GGGCGAACCAAAAAGAATGAT- G-3’、YGDN1:5’-CTTTTGTAGCCAATCATTTAT- CGGG-3’;YGUP2:5’-GGTTATGGAAGAAGG- AACCGAGAA-3’、YGDN2:5’-GCTATTTCCTC-TGCTTGTATTGGT-3’;YGUP3:5’-CTATTTTA- CGTCTTTGCGCGC-3’、YGDN3:5’-CCGAGCT- CGGGTTATGGAAG-3’;YGUP4:5’-CTGTAGA- CCCACGGAAAAATGT-3’、YGDN4:5’-GGTA- GAGCCGGATCGAAGT-3’。
Eppendorf管中,依次加入15 μL ddH2O、2.0 μL 10×缓冲液、0.4 μL dNTPs(10 mmol/L)、0.3 μL的上游引物(20 μmol/L)、0.3 μL下游引物(20 μmol/L)、1.0 μL DNA模板(50 ng/μL)和0.5 μLDNA聚合酶(2 U/μL)。PCR反应在伯乐C1000型PCR仪上进行,程序为:94 ℃变性5 min;32个循环中,94 ℃、30 s,54.3 ℃、45 s,72 ℃、100 s,共32个循环。PCR产物经电泳、割胶、回收和纯化后,将其与p-GEM T-easy载体(Promega)连接,置于4 ℃过夜。把连接产物导入大肠杆菌DH5α感受态细胞,经PCR检测后,各取3份菌液测序。
2.5 系统发育分析
从NCBI数据库中下载6条叶绿体基因组的序列,它们分别来自溪荪Donn ex Horn.(KT626943)、龙须菜Kunth(KX790361)、蔻第百子莲F. M. Leight(KX790363)、惠普尔丝兰(Torr.) Trel.、鹿葱Maxim.(NC_040164)和马褂木(Hemsl.) Sargent.(KU170538)。其中的溪荪为鸢尾科植物;蔻第百子莲和鹿葱属石蒜科(Amaryllidaceae);龙须菜和惠普尔丝兰为百合科(Liliaceae)植物;而外类群马褂木隶属木兰科(Magnoliaceae)。
利用Geneious 11.1.5内置的Find repeats工具预测叶绿体基因组的反向重复(inverted repeat,IR)序列、大单拷贝区(large single copy,LSC)和小单拷贝区(small single copy,SSC),并用EXCEL计算各自的GC值。Geneious 11.1.5软件中的MAFFT 7.388程序用于叶绿体基因组的多重比对,导出的比对序列用PhyML-SMS(Smart Model Selection in PhyML)在线工具获得最佳替代模型,最后生成最大似然(maximum likelihood,ML)树[25-26]。利用Mega X软件构建最大简约(maximum parsimony)树,自举检测值为1000。
3 结果与分析
3.1 叶绿体基因组的边界验证
利用NOVOPlasty对射干叶片DNA的Clean reads进行拼接,并通过PCR手段对4个边界进行克隆和测序验证。电泳结果表明,4对引物均能扩增出单1条带,PCR产物分别位于800、1300、1800、1300 bp处,大小与预期一致(图1)。序列测定结果表明,4条序列的长度分别为812、1328、1859、1325 bp,碱基排列与组装完成的叶绿体基因组边界序列完全一致。
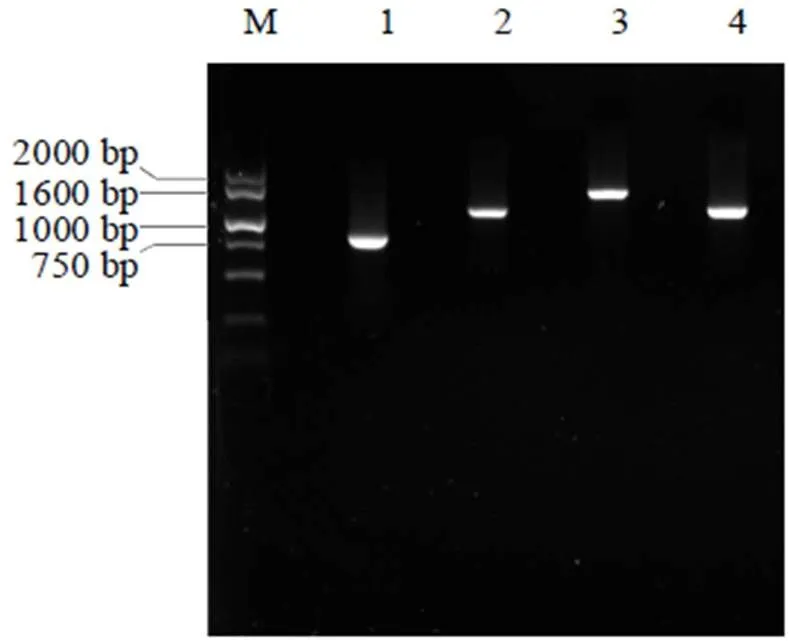
M-Marker 1~4-LSC/IRb、IRB/SSC、SSC/IRa和IRa/LSC边界序列
3.2 叶绿体基因组的特点
射干叶绿体基因组全长为153 816 bp,具有一个典型的四分体结构,即分别由LSC、SSC、IRa和IRb 4个部分组成(图2)。LSC与SSC的大小为83 143 bp和18 245 bp,IRa和IRb长度均为26 214 bp(表1)。利用Excel计算GC值,结果表明,反向重复区的GC值最大,达43.0%,LSC次之,为36.0%,而SSC的GC值最小,仅31.4%;射干整个叶绿体基因组的GC值为37.8%。
3.3 叶绿体基因的组成和特点
射干叶绿体基因组上共有133个基因,包括38个转运RNA(tRNA)、8个核糖体RNA(rRNA)、14个核糖体蛋白小亚基基因、11个核糖体蛋白大亚基基因、4个RNA聚合酶基因、12个NADH脱氢酶亚基基因、20个光系统I/光系统II亚基基因、6个细胞色素b/f复合物亚基基因和6个ATP合成酶亚基基因等。另有7个未知功能基因,它们是、、和,除外,其它各有2份拷贝。
rRNA基因中,、、和各有2份拷贝,分别位于2个反向重复区域,4种rRNA的长度分别为103、121、1491、2810 bp。tRNA中,、、、、、、和各有2份拷贝,其余tRNA均只有一个。具有2份拷贝的基因还有、、、、和基因等,也有2份拷贝,但其中1个为假基因(表2)。射干叶绿体基因组中,大部分基因没有内含子,少部分具1~2个内含子。、和基因有2个内含子,、、、、、、、、、、、、和具1个内含子(表2)。

内圈深色部分为GC含量
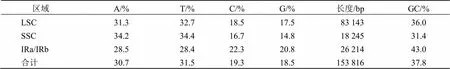
表1 射干叶绿体基因组的碱基组成
3.4 基因组特征比较分析
从NCBI下载了溪荪、龙须菜、蔻第百子莲、惠普尔丝兰、马褂木和鹿葱6种植物的叶绿体基因组,它们的全长分别为152 408、156 875、157 055、157 832、159 429和158 459 bp。马褂木叶绿体基因组的GC值最大,为39.2%,溪荪次之,为38.0%,蔻第百子莲的GC值最小,仅37.5%(表3)。马褂木的LSC和SSC最长,分别为87 766、18 997 bp,但它的IR较短,为26 333 bp。蔻第百子莲的IR最长,长度为26 869 bp,鹿葱次之,为26 764 bp,而溪荪的IR最短,仅26 026 bp。
射干与其他6种植物的LSC/IRb、IRb/SSC、SSC/IRa和IRa/LSC边界及基因分布如图3所示,基因在边界的排列情况基本相同。除马褂木外,LSC/IRb边界两侧分布有和基因,而马褂木LSC/IRb两侧为和基因。和基因位于IRb/SSC的边界,7种植物在该位置的基因全长为899~1112 bp,显著短于正常的基因,它们均为假基因。另一个位于SSC/IRa交界处,长度为5276~5489 bp,均为正常基因。马褂木IRa/LSC边界两侧分布有和基因,而其他6种植物在该处为和基因。

表2 射干叶绿体基因组的基因
×2-2份拷贝 Ψ-假基因*-一个内含子**-2个内含子
×2-2 copies Ψ-pseudogene*-one intron**-2 introns
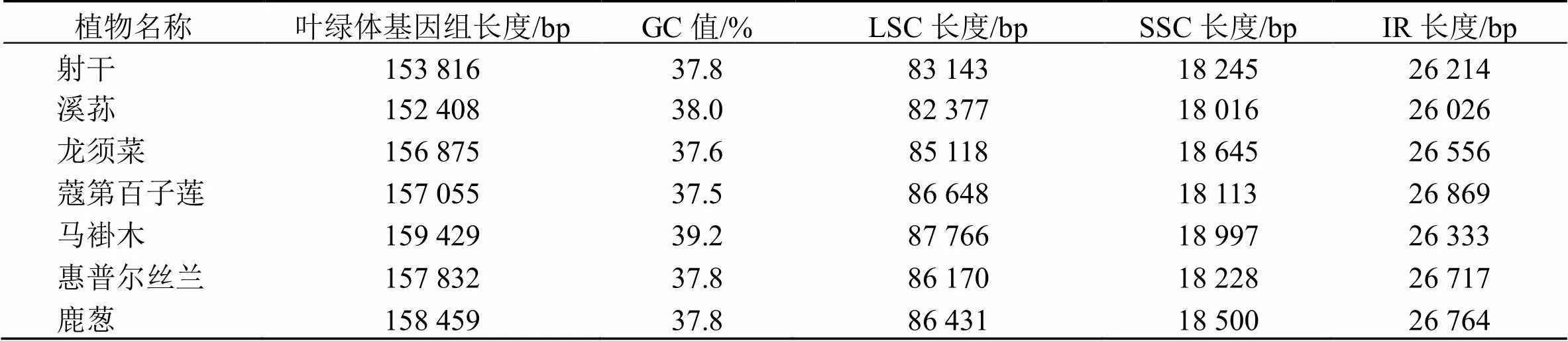
表3 7种植物叶绿体基因组的特征
3.5 系统发育分析
利用在线工具PhyML-SMS获得最佳分子进化模型,结果表明,7条叶绿体基因组的最佳模型为GTR+G+I,该模型的赤池信息标准(Akaike information criterion,AIC)与贝叶斯信息标准(Bayesian information criterion,BIC)分别为820 620.543 72和820 831.677 20,与其他模型相比,它们的数值最低。以马褂木叶绿体基因组为外类群,构建系统发育树(图4)。7种植物叶绿体基因组在系统发育树上可分为4组,石蒜科的鹿葱和蔻第百子莲聚于组I,支持率为100%;百合科的龙须菜和惠普尔丝兰聚于组II,支持率达100%;鸢尾科的溪荪和射干聚于组III,支持率也为100%;而外类群马褂木单独处于分支IV。同时,利用Mega X构建了最大简约树,结果与最大似然树基本一致,除龙须菜和惠普尔丝兰的支持率为99%外,其余均达100%(图4)。
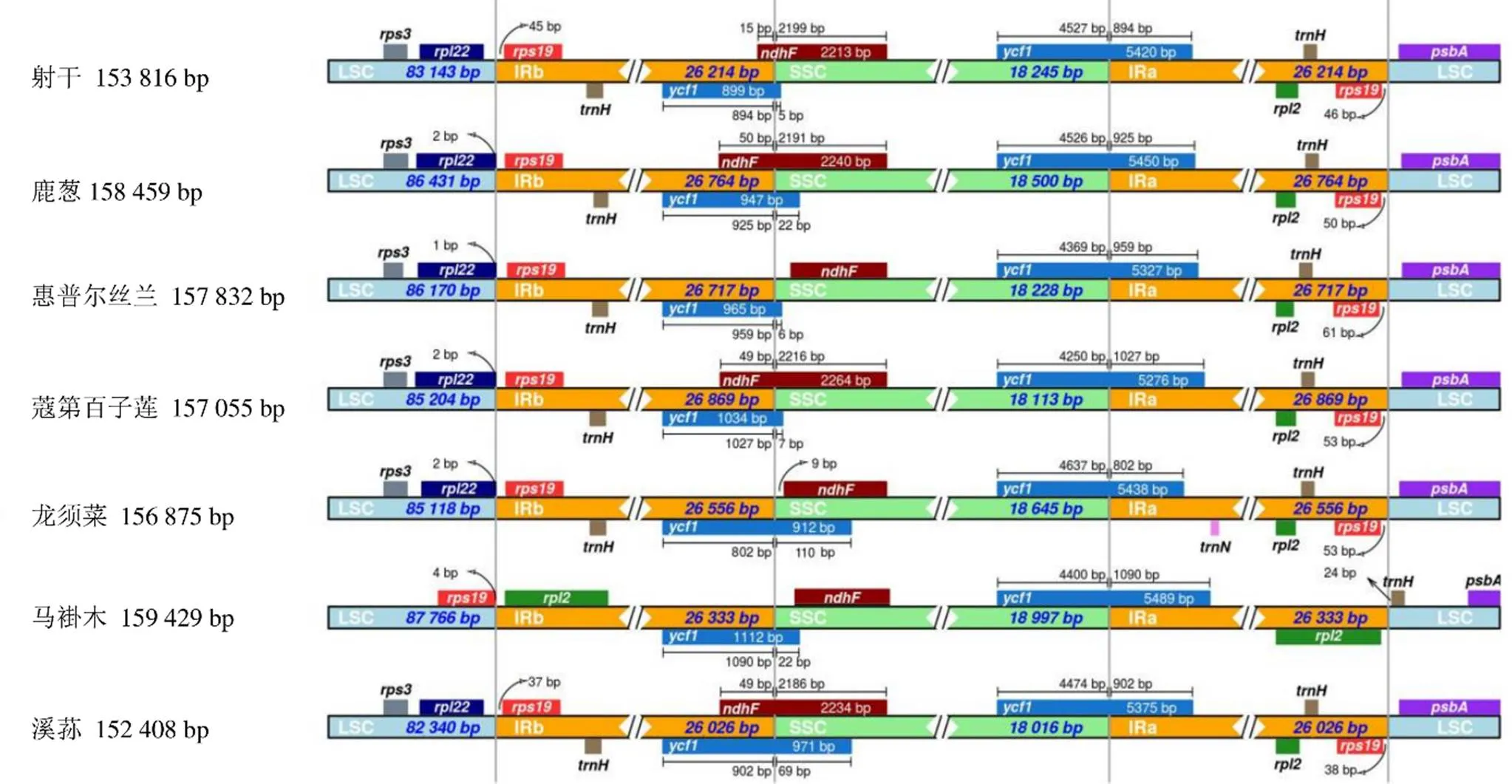
图3 射干叶绿体基因组的反向重复区域/单拷贝区域边界

图4 基于叶绿体全基因组构建的系统发育树
4 讨论
被子植物叶绿体基因组的结构通常十分保守,为双链环状,具2个反向重复序列IRa和IRb,它们将LSC和SSC隔开,最终形成四分体结构[27]。叶绿体基因组的长度也十分保守,大部分陆生植物的叶绿体基因组大小在135~160 kb[28]。禾本科(Gramineae)植物剪股颖L.的叶绿体基因组大小为136 584 bp[29];百合科洋葱L.的叶绿体基因大小为153 538 bp[30]。有些植物的叶绿体基因组较小,如药用植物木贼麻黄Bge.仅109 518 bp[31];而天竺葵Bailey的叶绿体基因组大小达217~942 bp,在目前已完成测序的陆生植物中最大[32]。本研究以射干为材料,在高通量测序的基础上,对其叶绿体基因组进行了组装,经PCR克隆和测序验证,得到射干叶绿体基因组的全长序列,它的长度为153 816 bp。
叶绿体基因组的基因组成十分保守,通常包含130个左右的基因,这些基因的功能涉及光合作用、转录和翻译等[33]。叶绿体基因组通常有114种基因,包括4种rRNA基因、30种tRNA基因和80种蛋白质编码基因[34]。在一些寄生或半寄生植物中,基因丢失现象十分普遍。山毛榉寄生(L.) W. P. C. Barton的叶绿体基因组大小仅70 kb,大部分基因丢失,仅剩下42个,与光合作用和叶绿体呼吸相关的基因全部缺失[35]。在半寄生植物广寄生(DC) Danser和桑寄生(Lecomte) Danser叶绿体基因组中,仅注释到106个基因,包括66个蛋白编码基因、28个tRNA、8个rRNA和4个假基因[36]。在射干叶绿体基因组中,共有4种rRNA基因,它们各有2份拷贝,分布在不同的IR区域,另有30种tRNA基因,其中的、和等各有2份拷贝。
植物在进化过程中,叶绿体基因组LSC/IRb、IRb/SSC、SSC/IRa和IRa/LSC边界常发生扩张或收缩事件,使叶绿体基因组大小出现一定的差异,甚至发生假基因化。比较鲁桑Perr.、蒙桑Schneid.、印度桑L.和川桑Schneid.的叶绿体基因组发现,它们的SSC/IRb交界处均存在1个假基因;在鼠尾草Thunb. SSC/IRb处的也是一个假基因。本研究中,也发现类似的现象,在射干叶绿体基因组中共有2份拷贝,位于SSC/IRb边界的为假基因,而另一份正常。叶绿体基因组可用于植物的进化分析和亲缘关系鉴定,目前已应用于不同分类级别的系统进化分析。张慧等[37]利用15个野芝麻亚科(Lamioideae)65个共有叶绿体蛋白序列构建最大似然法树(maximum likelihood,ML),发现益母草(Laur.) S. Y. Hu和水苏属Linn.的亲缘关系较近,大部分节点的支持率达100%。射干与其他6种植物叶绿体基因组的系统发育分析结果表明,射干与溪荪的关系最为接近,支持率达100%。
射干种质资源十分丰富,但不同种源药材的质量参差不齐。叶绿体基因组的组装和序列分析,为开发高分辨率的遗传标记提供了依据,也为后续开展该植物的群体遗传学和遗传多样性研究奠定了基础。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国科学院中国植物志编辑委员会. 中国植物志 [第16(1)卷] [M]. 北京: 科学出版社, 1985: 26.
[2] 张婧涵, 张晓瑞, 李国信, 等. 线性回归色谱峰定位法在射干药材多组分同时测定中的应用 [J]. 药物分析杂志, 2014, 34(7): 1149-1155.
[3] 廉伟伟, 熊维政, 贾玉梅, 等. 射干及其方剂临床应用探讨 [J]. 中医学报, 2018, 33(11): 2184-2190.
[4] 王迪, 辛旭阳, 尤献民, 等. 射干效用演变探析 [J]. 辽宁中医药大学学报, 2015, 17(9): 67-69.
[5] 展锐, 焦正花, 王红丽, 等. 射干的药理作用研究概况 [J]. 甘肃中医, 2011, 24(1): 78-80.
[6] 王晓荣, 朱序弼, 刘金. 耐寒耐旱射干西部开发佳卉 [J]. 中国花卉盆景, 2002(9): 33.
[7] 王继东, 卢杉. 延庆区射干栽培技术 [J]. 农业科技通讯, 2019(1): 192-193.
[8] 燕晨宇, 张翔, 秦民坚. 射干真叶、花蕾诱导愈伤组织的研究 [J]. 中国野生植物资源, 2018, 37(5): 16-19.
[9] 莫雪梅, 焦明姚, 文永刚, 等. 射干叶枯病防治药剂筛选试验初探 [J]. 耕作与栽培, 2017(4): 36-37.
[10] Li J Y, Ni G, Li L,. New iridal-type triterpenoid derivatives with cytotoxic activities from[J]., 2019, 83: 20-28.
[11] Tian M, Zhang X, Zhu Y,. Global transcriptome analyses reveal differentially expressed genes of six organs and putative genes involved in (iso) flavonoid biosynthesis in[J]., 2018, 9: 1160.
[12] Suzuki N, Koussevitzky S, Mittler R,. ROS and redox signalling in the response of plants to abiotic stress [J]., 2012, 35(2): 259-270.
[13] Liu X M, Zhou Y L, Xiao J W,. Effects of chilling on the structure, function and development of chloroplasts [J]., 2018, 9: 1715.
[14] Miller G, Shulaev V, Mittler R. Reactive oxygen signaling and abiotic stress [J]., 2008, 133(3): 481-489.
[15] Du Y P, Bi Y, Yang F P,. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses [J]., 2017, 7(1): 5751.
[16] Xiong A S, Peng R H, Zhuang J,. Gene duplication, transfer, and evolution in the chloroplast genome [J]., 2009, 27(4): 340-347.
[17] Daniell H, Lin C S, Yu M,. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering [J]., 2016, 17(1): 134.
[18] Twyford A D, Ness R W. Strategies for complete plastid genome sequencing [J]., 2017, 17(5): 858-868.
[19] Patel R K, Jain M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data [J]., 2012, 7(2): e30619.
[20] Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data [J]., 2017, 45(4): e18.
[21] Wyman S K, Jansen R K, Boore J L. Automatic annotation of organellar genomes with DOGMA [J]., 2004, 20(17): 3252-3255.
[22] Lowe T M, Eddy S R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence [J]., 1997, 25(5): 955-964.
[23] Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences [J]., 2004, 32(1): 11-16.
[24] Lohse M, Drechsel O, Kahlau S,OrganellarGenomeDRAW: A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets [J]., 2013, 41: W575-W581.
[25] Katoh K, Standley D M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability [J]., 2013, 30(4): 772-780.
[26] Guindon S, Dufayard J F, Lefort V,. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0 [J]., 2010, 59(3): 307-321.
[27] Bendich A J. Circular chloroplast chromosomes: The grand illusion [J]., 2004, 16(7): 1661-1666.
[28] Raubeson L A, Peery R, Chumley T W,. Comparative chloroplast genomics: Analyses including new sequences from the angiospermsand[J]., 2007, 8: 174.
[29] Saski C, Lee S B, Fjellheim S,. Complete chloroplast genome sequences of Hordeum vulgare,and, and comparative analyses with other grass genomes [J]., 2007, 115(4): 571-590.
[30] von Kohn C, Kiełkowska A, Havey M J. Sequencing and annotation of the chloroplast DNAs and identification of polymorphisms distinguishing normal male-fertile and male-sterile cytoplasms of onion [J]., 2013, 56(12): 737-742.
[31] Wu C S, Lai Y T, Lin C P,. Evolution of reduced and compact chloroplast genomes (cpDNAs) in gnetophytes: Selection toward a lower-cost strategy [J]., 2009, 52(1): 115-124.
[32] Chumley T W, Palmer J D, Mower J P,. The complete chloroplast genome sequence of: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants [J]., 2006, 23(11): 2175-2190.
[33] Daniell H, Lee S B, Grevich J,. Complete chloroplast genome sequences of,and comparative analyses with other[J]., 2006, 112(8): 1503-1518.
[34] 宋菊, 龙月红, 林丽梅, 等. 五加科植物叶绿体基因组结构与进化分析 [J]. 中草药, 2017, 48(24): 5070-5075.
[35] 李巧丽, 延娜, 宋琼, 等. 鲁桑叶绿体基因组序列及特征分析 [J]. 植物学报, 2018, 53(1): 94-103.
[36] 何懿菡, 韩立敏, 刘玉萍, 等. 鼠尾草叶绿体基因组序列分析 [J]. 植物研究, 2017, 37(4): 572-578.
[37] 张慧, 何帅兵, 孔繁德, 等. 益母草叶绿体基因组序列与系统进化位置分析 [J]. 中医药信息, 2018, 35(4): 21-27.
Structure, sequence characteristics, and phylogenetic evolution analysis ofchloroplast genome
JIANG Ming1, WANG Jun-feng2, ZHU Yan1, HUANG Wen-xin1, YING Meng-hao1, MA Jia-ying1, DAI Chen-yu1
1. College of Life Science, Taizhou University, Taizhou 318000, China 2. Scientific Research Management Center, East China Medicinal Botanical Garden, Lishui 323000, China
To confirm the genome structure, sequence characteristics, and phylogenetic relationship by sequencing and assembling the chloroplast genome of a medicinal plant.A PE150 strategy was applied to construct library. The complete chloroplast genome was generated using NOVOPlasty, followed by PCR confirmation of the borders, and sequence analysis, as well as phylogenetic study was conducted by bioinformatic tools.The full-length chloroplast genome was 153 816 bp in length, with a large single copy of 83 143 bp, an inverted repeat of 26 214 bp, and a small single copy of 18 245 bp. Thechloroplast genome consisted of 133 genes, including 92 protein-coding genes, 38 tRNAs, and 8 rRNAs, respectively. There were twogenes, one of which was a pseudogene. Phylogenetic evolution analysis results indicated that the seven chloroplast genomes can be divided into four groups,andfrom Iridaceae were found to cluster in the same clade, with a support rate of 100%.Assembly, sequence analysis and phylogenetic evolution ofchloroplast genome provides an insight into studies on both genetic structure and genetic diversity.
(L.) Redouté; chloroplast genome; structure; sequence analysis; Iridaceae
R282.12
A
0253 - 2670(2021)13 - 4039 - 08
10.7501/j.issn.0253-2670.2021.13.027
2020-12-03
台州市211人才工程经费资助(2012年度)
蒋 明(1973—),男,浙江嵊州人,博士,教授,硕士生导师,研究方向为植物基因组学、植物逆境生物学及其分子调控。E-mail: jiangming1973@139.com
[责任编辑 时圣明]
猜你喜欢
中国生殖健康(2018年1期)2018-11-06
天然产物研究与开发(2018年8期)2018-09-10
基层中医药(2018年5期)2018-08-31
天然产物研究与开发(2018年2期)2018-04-04
广西林业科学(2016年1期)2016-03-20
计算机与网络(2015年12期)2015-06-21
中国中医药现代远程教育(2014年14期)2014-03-01
食品科学(2013年6期)2013-03-11
中国烟草学报(2012年2期)2012-04-09
中国科技信息(2011年12期)2011-02-17