
食管癌化疗抗性的分子机制
2021-07-03 09:24许丽艳衡晶华李恩民
食管疾病 2021年2期
许丽艳,衡晶华,李恩民
目前,治疗食管癌的化疗药物主要是铂类和氟尿嘧啶类。在患者一般状况较好的情况下,也可使用紫杉醇或阿霉素等[1]。不同的化疗药物有不同的作用机理。顺铂(Cisplatin,DDP)通过与DNA分子中的嘌呤碱基上的N7反应,形成加合物,破坏细胞DNA的结构,干扰DNA修复,诱导癌细胞凋亡[2]。5-氟尿嘧啶(5-fluorouracil,5-FU)的细胞毒性机制归因于5-FU误掺入RNA和DNA分子中,以及对胸苷酸合成酶(thymidylate synthase,TS)的抑制。5-FU在细胞内转化为几种活性代谢物:氟脱氧尿苷一磷酸酯(fluorodeoxyuridine monophosphate,FdUMP)、氟脱氧尿苷三磷酸(fluorodeoxyuridine triphosphate,FdUTP)和氟尿嘧啶三磷酸(fluorouridine triphosphate,FUTP),它们均可破坏RNA和DNA的合成,以及抑制TS。5-FU分解代谢的限速酶是二氢嘧啶脱氢酶(dihydropyrimidine dehydrogenase,DPD),可将5-FU转化为二氢氟尿嘧啶(dihydrofluorouracil,DHFU)[3]。紫杉醇通过促进微管蛋白的聚合,阻止其解聚,发挥稳定微管的作用,抑制细胞微管的动态重组,将细胞周期阻滞于G2/M期,阻碍细胞分裂,诱发细胞死亡[4]。然而,化疗药治疗食管癌会产生各种耐药,导致对药物的应答率降低。本文从靶前耐药、靶上耐药和靶后耐药等不同角度,阐述细胞对DDP、5-FU以及紫杉醇的抗性机制。
1 靶前耐药
ABC(ATP binding cassette)转运蛋白家族,主要包括ABCB1基因编码的MDR1(multidrug resistance 1),又称P-糖蛋白或P-gp(P-glycoprotein),以及MRP1(multidrug resistance associated protein 1)和ABCG2(ATP-binding cassette subfamily G member 2)等[5]。ABC家族主要通过增强药物外排,降低细胞核内DDP浓度,导致耐药。SRPX2基因和FZD-7基因所编码蛋白均还可通过Wnt/β-catenin信号通路促进MDR1表达,促进食管鳞状细胞癌(ESCC)细胞的增殖和侵袭转移[6-7]。换言之,通过抑制MDR1可以逆转DDP抗性。例如YB-1(Y-box-binding protein 1)可抑制MDR1的转录,增强DDP诱导的EC109和TE-1细胞的细胞毒作用[8];恩替司他通过抑制Src/Mcl-1/MDR1信号轴,显著抑制DDP耐药小鼠的肿瘤生长[9]。从中草药“汉防己”中分离出来的汉防己碱(tetrandrine,TET)可抑制MDR1的表达和活性,干预耐药的发生[10]。另外,在TE-5R内发现DPD过表达导致5-FU的快速降解,DPD抑制剂吉美拉西可显著增加TE-5R细胞内5-FU浓度,减弱5-FU耐药性[11]。THUMPD2基因下调表达引起药物外排增多,进而导致DDP和5-FU抵抗[12]。相关情况见图1,ABC蛋白家族促进药物外排,降低核内药物浓度。
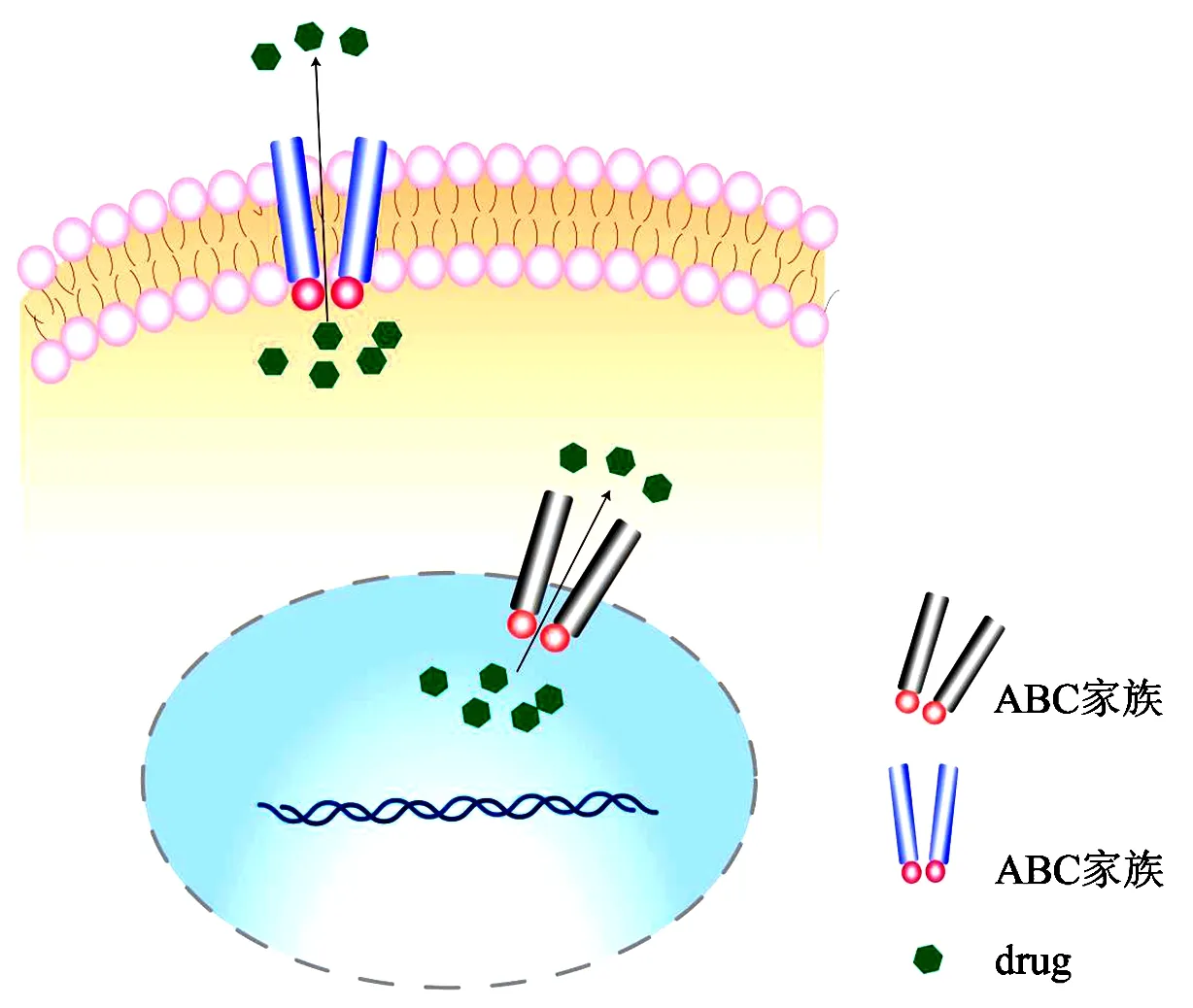
图1 靶前耐药模式图
2 靶上耐药
2.1 DNA损伤修复介导DDP抗性
如果DNA损伤发生在有丝分裂前,并且无法修复,就会诱发细胞凋亡。因此,DNA损伤修复(DNA damage repair,DDR)是肿瘤细胞逃避因化疗药物毒性引起细胞死亡的重要的耐药机制。
2.1.1 DNA损伤修复途径损伤修复方式包括碱基切除修复(base excision repair,BER)、错配修复(mismatch repair,MMR)、同源重组(Homologous Recombination,HR)、非同源末端连接(nonhomologous end joining,NHEJ)等。①BER:铂类化合物诱导细胞产生的活性氧自由基(reactive oxygen species,ROS),可导致脂质、蛋白质、胞核DNA和线粒体DNA(mitochondrial DNA,mtDNA)损伤。其中,最稳定且有害的ROS修饰产物之一是8-羟基鸟嘌呤(8-Hydroxyguanine,8-oxoG),该损伤修复通过BER进行。其中,hOGG1-1a(a-hOGG1)主要修复核DNA损伤,hOGG1-2a(b-hOGG1)主要修复mtDNA损伤,介导DDP抗性[13]。②MMR:XPC是一种重要的DNA损伤识别蛋白,它不仅参与核苷酸切除修复(nucleotide excision repair,NER),还参与MMR和双链断裂修复途径[14]。③HR修复:增强细胞损伤修复能力的BRCA1作为HR系统的关键功能蛋白,可以通过与BARD1形成异源二聚体参与DDR。整合素α-5通过粘着斑激酶FAK激活PI3K/AKT信号通路,作用于BARD1,激活DDR,抗细胞凋亡,介导DDP耐药[15]。④NHEJ修复:DNA依赖的蛋白激酶全酶(Ku70、Ku80和DNA-PKcs)是NHEJ的关键启动酶。HOXB7使Ku70、Ku80和DNA-PKcs表达上调,将细胞周期阻滞于S期,产生DDP抗性[16]。
2.1.2 ATM和ATRATM(ataxia-telangiectasia mutated)和ATR(ataxia telangiectasia and Rad3-related)的激活对于DDR和维持基因组稳定性至关重要,抑制ATM或ATR被认为是一种很有前景的化疗增敏策略。E2F1激活miR-26b的转录,其与ATM和RB基因的3UTR相互作用,下调ATM和RB的表达,增强DDP对食管癌化疗的敏感作用[17]。STAT3对于有效修复受损的DNA也是必不可少的。ATR的阻断剂VE-822与DDP的联合作用显著抑制了食管癌KYSE450和KYSE70细胞中p-STAT3的表达,特别是对ATM缺陷型细胞的增敏作用更加明显,并且引起了包括Hippo通路、MAPK通路、JAK-STAT通路和PI3K-AKT通路的广泛变化[18]。
2.1.3 其他定量蛋白质组学结合生物信息学和蛋白功能分析发现,RIP3通过上调HSP90-CDC37复合物和ERK、JNK和AKT激酶等下游信号,以一种不依赖激酶的方式调节DNA修复,导致DDP靶向耐药[19]。
2.2 核苷酸代谢与5-FU抗性
核苷酸代谢中的关键酶如DPD上调表达和TS活性增加,参与了癌细胞对5-FU耐药。ID-1与E2F1竞争结合Cdc20,激活IGF2基因表达,阻断IGF1R(Insulin-like growth factor type 1 receptor),间接上调TS,介导5-FU耐药[20]。miR-338-5p与ID-1编码基因的mRNA的3’-UTR直接相互作用,抑制ID-1表达,介导ESCC对5-FU增敏[21]。二甲双胍诱导核苷酸代谢改变,如其中TS和胸苷激酶TK1(thymidine kinase 1)的表达增加可能导致细胞内dTTP池的增加和5-FU合成代谢物的“稀释”,介导5-FU耐药[22]。胰岛素通过增加5-FU摄取,增加胞内游离TS、降低TS三元复合物,以及上调活性的Caspase-3的表达,促进对5-FU增敏[23]。细胞周期阻滞使胸腺嘧啶耗竭,介导5-FU增敏。REV3L表达下调通过抑制CyclinD1和Survivin的表达,诱导G1期阻滞和凋亡,增加细胞对5-FU的敏感性[24]。过表达miR-145,靶向REV3L,显著提高5-FU抑制抗凋亡经典分子Bcl-2家族蛋白表达,提高细胞凋亡率[25]。
2.3 抑制G2/M期阻滞介导紫杉醇耐药
在多个紫杉醇耐药的细胞系中,发现细胞周期分布改变:G0/G1和S期细胞增多,G2/M期细胞减少[26]。5-AZ可通过诱导细胞周期相位重新分布,使耐药ESCC细胞对紫杉醇化疗重新增敏[27-28]。STMN1(Stathmin)通过促进微管解聚和/或防止微管蛋白异二聚体聚合,调节微管动力学;沉默STMN1基因表达可以通过G2/M期阻滞增加ESCC对紫杉醇的敏感性[29-30]。有关食管癌化疗紫杉醇和5-FU的靶上耐药机制见图2,其中DNA损伤修复介导DDP抗性;TS、TK1增加胞内dTTP池,降低5-FU合成代谢物浓度,介导5-FU抗性;解除G2检查点介导紫杉醇抗性。
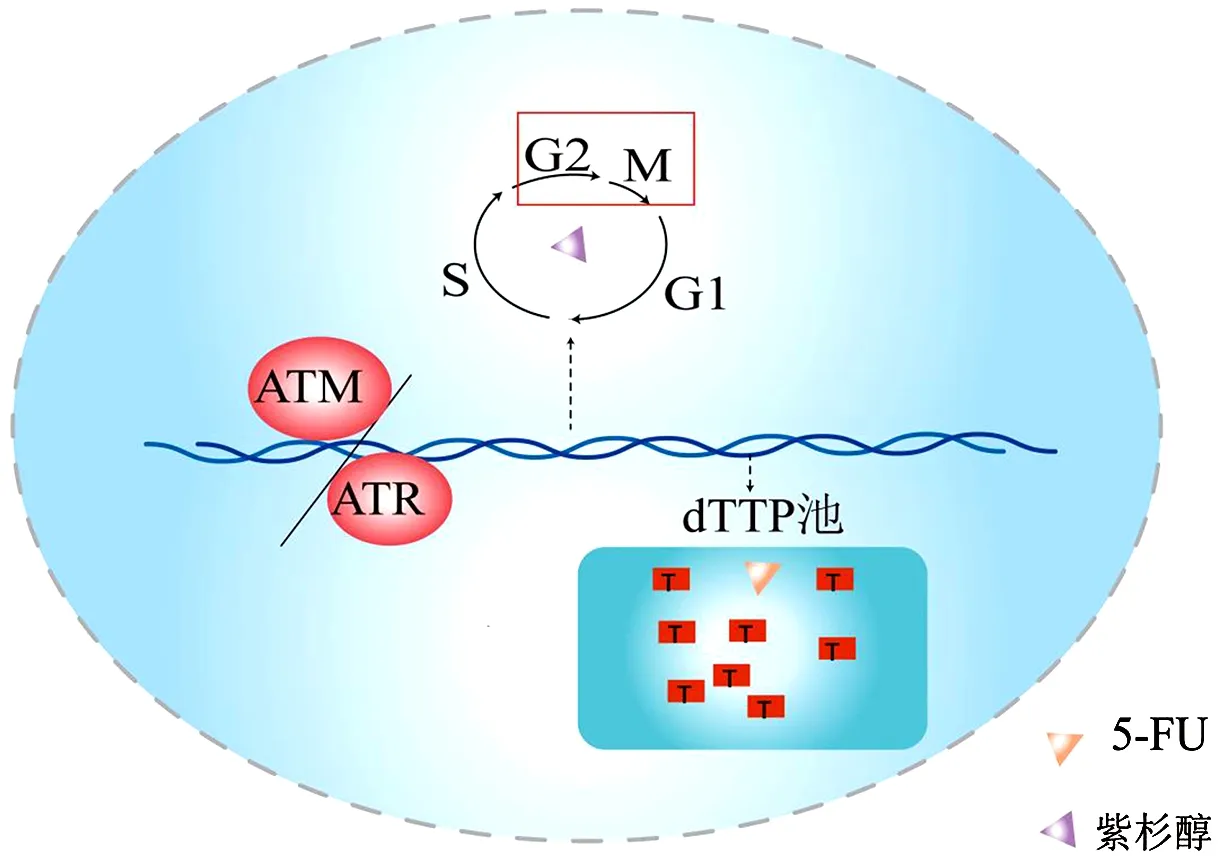
图2 靶上耐药模式图
3 靶后耐药
肿瘤细胞可以通过细胞程序性死亡系统的改变对化疗药物产生抵抗,涉及肿瘤细胞本身及肿瘤微环境(tumor microenvironment,TME)。有关食管癌靶后耐药相关机制见图3,促进增殖、抑制凋亡、侵袭转移、肿瘤细胞干性、Warburg效应、CAFs分泌因子均介导不同化疗药物耐药。
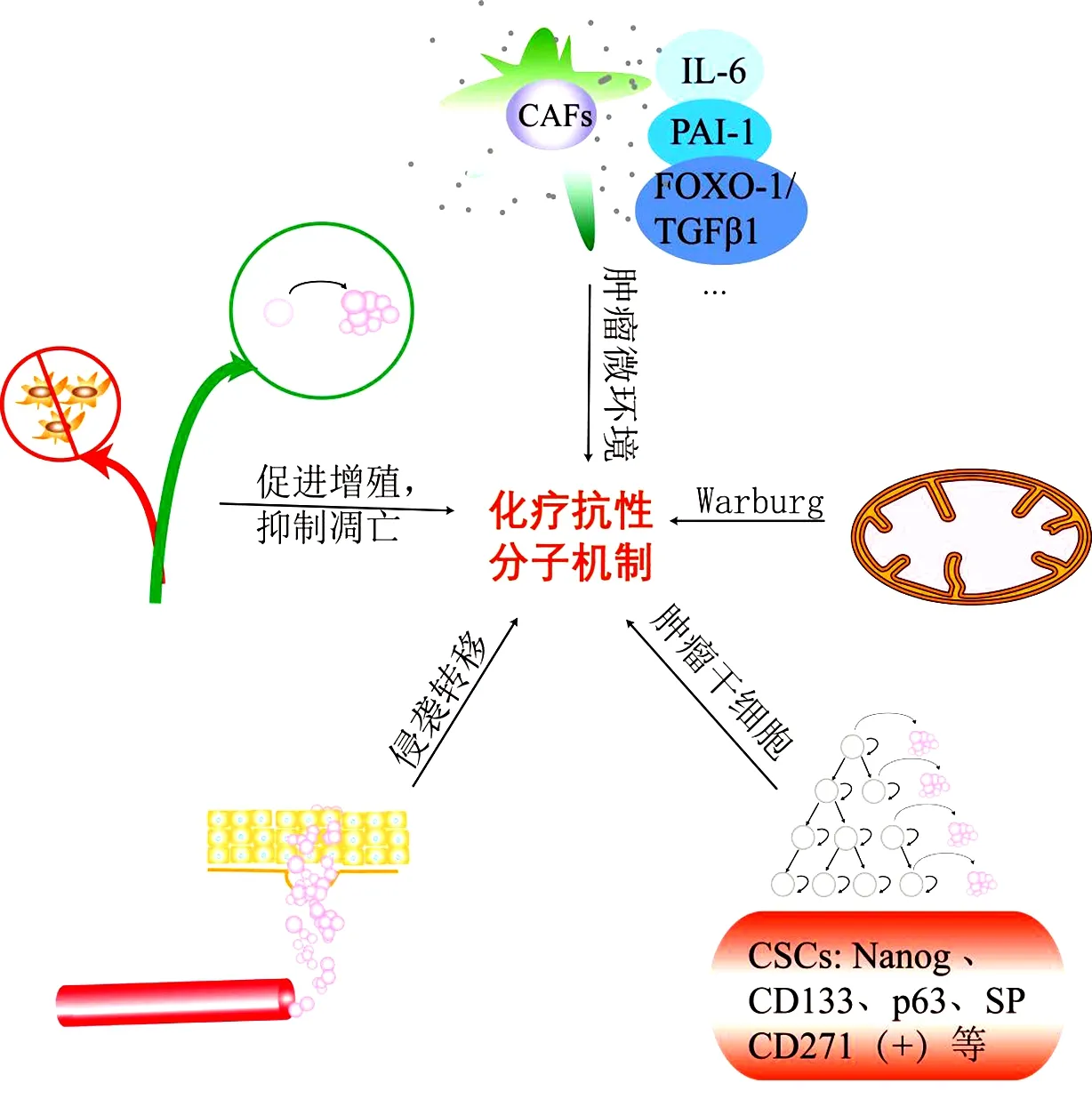
图3 靶后耐药模式图
3.1 抑制细胞凋亡、促进细胞增殖、介导化疗抗性
3.1.1 凋亡抑制蛋白家族介导DDP抗性凋亡抑制蛋白(inhibitor of apoptosis proteins,IAPs)的抗细胞凋亡能力在化疗抗性中发挥重要作用。Survivin是细胞凋亡抑制蛋白IAPs家族的成员,能够抑制半胱天冬蛋白酶,阻断细胞死亡[31]。miR-214-3p靶向下调Survivin和CUG-BP1表达,诱发ESCC细胞化疗增敏[32]。Caspase激活因子Smac通过拮抗IAPs,促进细胞凋亡。Smac基因敲除显著抑制DDP诱导的线粒体膜电位塌陷、Caspase激活和细胞色素c释放所引起的细胞凋亡,导致体内外DDP耐药[33]。XIAP(X chromosome-linked inhibitor of apoptosis)是IAPs家族的另一成员,靶向抑制Caspase 3和Caspase 9。HSP90(heat shock protein 90)抑制剂17-AAG和DDP联用,通过抑制AKT/XIAP,激活Caspase-3,PARP降解,诱导细胞凋亡,介导DDP的增敏[34]。
3.1.2 PI3K/AKT/mTOR途径激活介导化疗抗性PI3K/AKT/mTOR信号通路在调控细胞生长、存活和凋亡等过程中发挥重要作用。AKT将BAD蛋白136丝氨酸残基位点磷酸化,降低其与Bcl-2的亲和力,导致抵抗细胞凋亡[35]。PPP2R1B基因编码的磷酸酶PP2A可以使AKT去磷酸化,增强化疗的敏感性,而miR-200C靶向抑制PPP2R1B表达,增强对DDP的抗性[36]。抑制PI3K/AKT/mTOR信号通路上游负性调控因子PTEN,增强化疗抗性。Cyclin B1通过PTEN/AKT途径,抑制Bcl-2依赖的线粒体调节的内源性死亡途径,产生抗性,且体外实验显示,miR-141-3p直接与PTEN的3’-UTR结合,抑制细胞凋亡,促进EC9706R细胞获得耐药[37,38]。mTOR介导化疗抗性。PP242能抑制mTORC1和mTORC2的活性以及mTORC1依赖的PI3K/AKT的反馈激活,促进DDP诱导的细胞凋亡[39]。
3.1.3 NF-kB介导化疗抗性在肿瘤细胞中,NF-κB的表达可能是影响以5-FU为主的化疗的关键因素[40]。siRNA抑制NF-κB蛋白表达可以增强5-FU的抗细胞增殖作用[41]。p65 的siRNA抑制NF-κB的活化,提高针对5-FU的裸鼠模型的化疗敏感性[42]。然而,RNAi方法作用时间短,极大地限制了其临床应用。在ESCC和EAC中均发现,姜黄素通过抑制IkBa磷酸化,抑制NF-kB的活化,下调Bcl-2和CyclinD1,使细胞周期阻滞,增加细胞凋亡率[43-44]。
3.1.4 p38与p53以不同方式影响化疗p38蛋白作为MAPK家族中的成员与诱导凋亡相关。miR-29c通过直接靶向抑制FBXO31基因表达和p38蛋白激活,在体内外诱导细胞凋亡,抑制细胞增殖,逆转5-FU的化疗耐药性;而STAT5A直接与miR-29c启动子结合抑制其转录,产生相反的效应[45]。与p38δ阳性的ESCC相比,p38δ阴性的ESCC对DDP和5-FU耐受性更强[46]。P53基因在70%的EAC中发生突变,导致化疗耐药。突变型P53基因的异位表达使P53基因缺失的细胞对APR-246敏感,而P53基因敲除或敲降降低了药物活性。APR-246作为突变型P53复活剂在CLX和PDX模型中显示了强大的抗肿瘤活性,并恢复了对DDP/5-FU耐药异种移植模型的化疗敏感性[47]。
3.1.5 其他靶点分子及通路lncRNA CCAT1在体内外通过调节miR-143/PLK1/BUBR1信号轴,促进细胞增殖,增强DDP耐药性[48]。BMI1和Mel18共同抑制c-Myc,增加细胞凋亡和细胞毒性,介导对DDP的增敏[49]。在补充甲基硒酸的EAC细胞中,硒结合蛋白SELENBP1稳定过表达,导致细胞凋亡,细胞衰老和DDP细胞毒性增加[50]。在EAC细胞及小鼠模型中,Deferasirox和DFO作为铁螯合剂有效地抑制了细胞对铁的摄取,促进细胞内铁的动员,降低了细胞活力和增殖,增强DDP、5-FU和阿霉素对细胞的毒性[51]。
3.2 肿瘤干细胞介导DDP抗性
肿瘤干细胞(Cancer stem cells,CSCs)或肿瘤起始细胞(tumor initiating cells,TICs)在肿瘤的发生、转移和复发中发挥主导作用,同时也可能是化疗失败的源头。①干细胞标志物与耐药:Nanog蛋白决定胚胎发育过程中多能内细胞团(inner cell mass,ICM)的命运,介导DDP抵抗[52]。CD271+、整合素α6bri/CD71dim和/或过表达Achaete-Scute复合物同源物2似乎至少代表一个干细胞亚群,抑制对5-FU和DDP的敏感性[53-54]。②抑制干细胞分子标志物介导化疗药物增敏:IGF2的中和抗体,通过IGF2-PI3K/AKT-miR-377-CD133信号轴,抑制肿瘤干性,增强裸鼠移植瘤对5-FU治疗的敏感性[55]。塞来昔布通过抑制COX2表达,减少CSC分子标志物,使食管癌细胞对5-FU敏感[56]。低浓度的全反式维甲酸(all-trans retinoic acid,ATRA)与5-FU和DDP联合使用,可促进CSCs凋亡,以及G2/M和G0/G1期细胞周期阻滞,增强DDP和5-FU的细胞毒作用[57]。③侵袭与耐药:干细胞相关蛋白,Nanog、p63和Bmi-1,以及EMT相关蛋白,N-cadherin和纤维粘连蛋白,在P75NTR阳性组中表达水平显著增高,对DDP的抗性也显著增高[58]。BMP结合卵泡抑素样蛋白FSTL1,通过典型的NFκB途径激活和BMP途径减弱,在ESCC的发生和转移中发挥重要的癌蛋白的作用,介导DDP抗性[59]。染料外排亚群(side population,SP)细胞作为人食管癌干细胞(esophageal cancer stem cells,ECSCs)样亚群的一个关键特征,对化疗的抵抗力显著高于大多数其他癌细胞。长期应用化疗可能通过SP细胞的富集,使原本对化疗敏感的食管癌产生获得性耐药,促进癌细胞向远处转移[60]。④miRNAs与耐药或增敏:如let7、miR-134、miR-296、miR-302、miR-367和miR-470,参与干细胞功能调节,如自我更新、多能性和分化。沉默miR-455-3p使包括Wnt/β-catenin和TGF-β等多条TIC相关通路失活,减少CD90+和CD271+TIC亚群,可使ESCC细胞对DDP增敏[61]。
3.3 TME改变调控DDP抗性
化疗耐药性是一种复杂的多因素参与的过程,同癌细胞与TME之间的相互作用有关。越来越多的证据表明癌细胞、成纤维细胞、内皮细胞、巨噬细胞和ECM成分组成的TME在促进实体肿瘤对化疗耐药的发生中发挥着关键作用。IL-6通过激活STAT3/NF-κB信号通路,上调CXCR7的表达,介导DDP抗性[62]。涉及癌症相关成纤维细胞(cancer-associated fibroblasts,CAF)的化疗耐药机制包括调控癌细胞与ECM相互作用的通路、CAF-ECM黏附和细胞因子或趋化因子介导的信号转导。在肿瘤进展的早期阶段,TGFβ1与其受体的胞外区结合抑制癌细胞的增殖,在晚期促进上皮向间充质转化和转移。CAF可通过FOXO-1/TGFβ1信号环介导DDP以及紫杉醇耐药[63]。PAI-1作为DDP刺激CAF的旁分泌因子可以通过激活AKT/ERK,影响体外培养的食管癌细胞增殖和耐药,并且PAI-1使caspase和γH2AX失活,降低ESCC细胞的DNA损伤和ROS水平,抑制氧化应激,以保护肿瘤细胞免受DDP的影响[64]。
3.4 侵袭转移介导DDP抗性
激活后的SOX9通过与miR-203a启动子结合抑制其转录,阻止miR-203a对PI3K/AKT/mTOR的抑制作用,促进ESCC侵袭,介导DDP抗性[65]。miR-125a-5p的过表达显著降低STAT3和p-STAT3及其下游靶基因VEGF在ESCC细胞的蛋白水平,介导DDP增敏[66]。达沙替尼通过抑制PI3K/AKT和STAT3通路,显著提高DDP对ESCC细胞KYSE410的Caspase的激活,促进细胞凋亡,抑制ESCC细胞侵袭和血管生成[67]。DDK3处理的EAC细胞OE33可以通过TGF-β促进血管内皮形成,对5-FU和DDP的耐药性明显增强[68]。miR-221直接靶向抑制DKK2激活Wnt/β-catenin通路,促进EMT,最终导致化疗抵抗[69]。
3.5 代谢
在大多数情况下,癌细胞主要依赖有氧糖酵解提供能量和代谢物质。实体肿瘤组织中的缺氧状态不仅为ESCC细胞的生长与增殖创造了独特的环境,同时也使ESCC对包括化疗在内的常规癌症治疗的反应性减弱。Warburg效应调节因子,SLC2A1又称GLUT1,是主要的葡萄糖转运蛋白之一,可促进糖酵解效率增加,以满足细胞生长的需要。突变型P53的表达可促进GLUT1从细胞质到细胞表面的转位,增加葡萄糖的摄取。PAX5编码基因敲低可以通过PAX5-p53-SLC2A1轴促进细胞增殖,增强DDP抗性[70]。AMPK(AMP-activated protein kinase)是Warburg效应的负调节因子,其活性形式可下调能量消耗过程。水飞蓟宾可以激活AMPK,逆转Warburg表型,协同5-FU发挥抗癌作用[71]。ATP调节因子,ALC1是ATP依赖的染色质重塑酶,它通过抑制PI3K/AKT途径,抑制糖酵解,抑制细胞生长,增强DDP对ESCC细胞的细胞毒作用[72]。RAC1的沉默抑制AKT/FOXO3a信号转导,抑制细胞糖酵解,增敏DDP的治疗[73]。磷酸戊糖途径(pentose phosphate pathway,PPP)将葡萄糖代谢与核酸生物合成联系起来,提供谷胱甘肽还原酶所需的NADPH,产生用于ROS解毒的GSH。丙酮酸激酶是糖酵解中的限速酶,抑制丙酮酸激酶M2(pyruvate kinase M2,PKM2),减少葡萄糖流入PPP,恢复对DDP的敏感性,导致DDP治疗后细胞内ROS水平的增加[74]。YQ23作为一种新型的氧气载体,能抑制HIF-1α的表达,进而使肿瘤表型从较多的间质状态转变为上皮状态,逆转对DDP和5-FU的抗性[75]。
受限的葡萄糖水平已被证明可以诱导细胞周期停滞,减缓细胞增殖速度,使肿瘤细胞进入静止状态。在缺糖条件下,糖酵解受限,二甲双胍处理的EC109细胞的能量供应受到明显限制,AKT磷酸化受到明显抑制,严重损害了细胞的DNA修复过程[76]。二甲双胍在体内外均能显著抑制4EBP1和S6K1的表达,促进细胞凋亡,增强DDP的作用[77]。
4 总结
顺铂、5-FU、紫杉醇的作用部位前中后的机制有相似之处。三者靶前耐药机制主要为膜转运蛋白ABC家族促进药物外排。靶上耐药机制则因药物本身作用部位不同而异,如顺铂与DNA损伤修复相关,5-FU与核苷酸代谢相关,紫杉醇则与G2M期相关。靶后耐药与增殖、凋亡、肿瘤干细胞、肿瘤微环境、侵袭转移以及代谢等相关。尽管耐药机制令人望而生畏,但不应忽视这样一个事实,即化疗在多数情况下都是有效的,显著延长了患者的生命,在某些情况下,还能治愈食管癌。因此,寻找化疗耐药或增敏的关键分子,减少化疗耐药,是现在以及未来的重点和难点。
猜你喜欢
空军军医大学学报(2022年2期)2022-11-24
当代水产(2022年7期)2022-09-20
西南农业学报(2022年3期)2022-04-25
科教新报(2022年10期)2022-03-27
家庭医药(2022年3期)2022-03-24
昆明医科大学学报(2021年12期)2021-12-30
现代仪器与医疗(2021年2期)2021-07-21
植物保护(2019年2期)2019-07-23
婚育与健康(2017年8期)2017-10-27
中国民族民间医药·下半月(2011年4期)2011-09-27
