
钯催化氧化N—H键羰基化反应合成1,3,4-噁二唑-2(3H)-酮杂环化合物机理的理论研究
2021-06-29 06:01李昌华薛珊珊张婷婷贾建峰武海顺
高等学校化学学报 2021年6期
任 颖,李昌华,王 涛,薛珊珊,张婷婷,贾建峰,武海顺
(山西师范大学化学与材料科学学院,磁性分子与磁信息材料教育部重点实验室,临汾 041004)
随着全球环境问题的不断凸显,寻求对环境友好和低能耗低排放的有机合成方法是现代化学家们的一致追求.在有机分子内引入一氧化碳(CO)合成具有碳碳键、碳杂键目标产物的羰基化反应,因其高选择性、符合绿色化学的理念而备受当代科学家的青睐.噁二唑酮类化合物是一类含有O,N原子组成的五元杂环结构,其中1,3,4-噁二唑-2(3H)-酮类化合物以其突出的杀虫、消炎、抗菌等生物活性在农药和医药领域有着非常广泛的应用价值.实验研究表明,1,3,4-噁二唑-2(3H)-酮类化合物的核心结构是许多抗肿瘤抗癌药物、农药、除草剂中的重要结构单元,因此受到了化学、生物、药物等领域的广泛关注[1~7].
近几十年来,已尝试多种方法合成1,3,4-噁二唑-2(3H)-酮类化合物及其衍生物,但是由于环氧丙烯、二氯化碳等化合物的高化学反应性及二氯化碳的高毒性使得这些合成方法无法实现[8,9].过渡金属催化的偶联反应已成为有机合成中一种简单、直接、有效的方法,钯催化的偶联反应具有反应条件温和、副产物少及易于处理等优点,已成为构筑碳碳键和碳杂键等的重要方法之一[10~15].2015 年,Jiang等[16]报道了以苯甲酰肼和一氧化碳作为反应物,在钯的催化作用下,实现了羰基和氨基的直接氧化O—H/N—H键羰基化反应,达到了合成1,3,4-噁二唑-2(3H)-酮类化合物的环保性和高效性(Scheme 1).

Scheme 1 Pd(TFA)2-catalyzed O—H/N—H carbonylation for the synthesis of 1,3,4-oxadiazole-2(3H)-one[16]
实验提出的反应机理如Scheme 2 所示.首先,三氟乙酸钯[Pd(TFA)2]与底物作用经过分步的N—H键活化形成中间体B.接着CO与中间体B的Pd原子配位并插入Pd─N键得到五元的金属环化合物C.随后,发生还原消除生成产物和零价钯催化剂.最后Pd(0)在CuO和三氟乙酸(TFAH)作用下使Pd(TFA)2催化剂再生,完成催化循环.

Scheme 2 Proposed mechanism for the Pd-catalyzed carbonylation[16]
虽然实验给出了可能的反应机理,但是仍有几个重要的科学问题需要进一步研究:(1)整个催化循环中具体的催化机制,(2)反应的决速步骤,(3)反应底物中不同N—H 键活化的竞争性.基于实验猜测的反应机理,采用密度泛函理论对钯催化氧化N—H 键羰基化反应合成1,3,4-噁二唑-2(3H)-酮进行了系统的理论研究,探究了配体效应和取代基效应对反应的影响,希望能够通过理论计算对反应机理有一个清楚的认识,以便对今后实验的改进和发展提供参考.
1 计算方法
所有计算均采用Gaussian 09程序完成[17].采用密度泛函理论M06方法对反应涉及的所有中间体和过渡态进行几何结构优化和能量计算[18,19].对Pd 原子采用基于有效核势能近似(ECP)的LANL2DZ 基组[20,21],对其它原子(C,H,O,N,F)采用6-31G(d,p)全电子基组[22,23].在相同的计算水平下对优化后的构型进行频率计算,确认势能面的极小值点是中间体(无虚频)或一级鞍点是过渡态(有且仅有一个虚频),并通过内禀反应坐标(IRC)计算分析[24,25],对过渡态进行验证.为了证明计算方法结果的可靠性,将优化后的结构在B3LYP/6-311+G(d,p)理论水平下进行单点能的计算[26~28].基于气相优化的几何结构,利用溶剂化模型SMD[29],在相同的计算水平下对优化后的结构进行单点能计算,溶剂为乙腈(Acetonitrile),文中涉及的能量均为溶剂化效应下的热力学校正的自由能.对于体系中的一些关键构型进行了自然键轨道(NBO)电荷布局分析[30].采用CYLview软件对优化的关键中间体和过渡态的结构进行了绘制[31].
2 结果与讨论
根据实验结果,以苯甲酰肼和CO 为反应物模型(Scheme 3),探究了在Pd(TFA)2的催化下生成1,3,4-噁二唑-2(3H)-酮的可能反应机制,具体讨论如下.

Scheme 3 Model reaction of Pd(TFA)2-catalyzed N—H carbonylation for the synthesis of 1,3,4-oxadiazole-2(3H)-one
2.1 反应机理
催化剂Pd(TFA)2中的Pd中心原子与反应底物络合时,可以是N1配位、N2配位或O配位,分别得到3种配合物异构体1a,1b和1c(图S1,见本文支持信息).计算结果表明,配合物1a,1b和1c的相对能量分别为-86.6,-32.0和-22.5 kJ/mol.可见配合物1a较其它两种配位要更稳定.同时也对苯甲酰肼底物进行了NBO 分析(图1),N1 原子、N2原子和O 原子的电荷分别为-0.715,-0.524和-0.608 e,可见,N1 原子的负电性强于O原子和N2 原子,即N1 原子的亲核性更强,所以N1 原子更易与Pd 原子配位.所以,只考虑了最稳定的异构体1a.
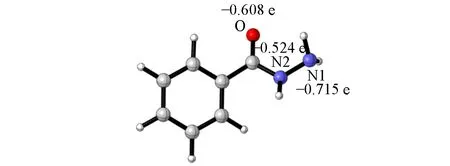
Fig.1 NBO charge population analysis on benzohydrazide
从配合物1a 开始,反应的第一步是N1—H 活化,涉及3 种可能的机理:协同金属化/去质子化(Concerted metalation/deprotonation,CMD)、σ-键复分解(σ-Bond metathesis,meta)和氧化加成(Oxidative addition,oxd),如图2所示.在CMD机理中,经过一个经典的六元环过渡态TS(1a/2)CMD,N1—H键断裂,O—H键形成得到中间体2CMD.1a→TS(1a/2)CMD→2CMD过程需克服30.7 kJ/mol的能垒.随后脱去一分子TFAH,底物中O与Pd金属中心配位形成五元金属环中间体3.在σ-键复分解机理中,通过一个四元环钯结构的过渡态TS(1a/2)meta,氢原子迁移至与Pd 配位的O 原子上,生成了中间体2meta.该过程(1a→TS(1a/2)meta→2meta)需要克服96.1 kJ/mol 的能垒.而在氧化加成机理中,经过一个三中心过渡态TS(1a/2)oxd氢原子迁移至Pd 金属中心,导致Pd 的氧化态从二价到四价,从而需要克服高达221.5 kJ/mol的能垒.鉴于此步的高能垒,在动力学上是不可行的,因此,并未进一步进行氧化加成路径的计算.对比N1—H活化步中的3种机理,CMD机理过渡态的相对能量分别比σ-键复分解和氧化加成机理的过渡态低65.4 和190.8 kJ/mol.所以,CMD 机理最优.需要指出的是,N—H 键活化的机理与文献[32]报道的C—H活化反应机理相类似.

Fig.2 Calculated Gibbs energy profiles for N1—H activation
根据反应步骤顺序的不同提出了两种反应机理:机理A(N2—H活化/CO插入/还原消除)和机理B(CO插入/N2—H活化/还原消除).
2.1.1 机理A(N2—H活化/CO插入/还原消除)根据实验[16]提出的反应机理(Scheme 2),反应的第2步是N2—H活化.基于中间体3的几何构型,在N2—H活化步骤中,考虑到N1—H和N2—H活化的竞争性,猜测了3种可能的路径(图3).路径1是一个“三步反应”,首先N1的H原子迁移到金属Pd中心上形成Pd—H 键,然后Pd—H 键断裂,H 原子进一步迁移到CF3COO-(TFA)配体的O 原子上,最后N2 的氢迁移到N1原子上完成N2—H活化.3→ATS(5/7)路径的能垒达到了318.5 kJ/mol.路径2是一个“两步反应”,经过渡态ATS(3/6)先发生N2与N1原子间的氢迁移活化N2—H键,接着已经迁移到N1原子上的H通过过渡态ATS(6/7)迁移到CF3COO-配体的O原子上,但这一过程需要克服较高的能垒(178.6 kJ/mol).另外,路径3是N2—H键直接活化的“一步反应”,即可实现N2原子与配体CF3COO-上O原子之间的氢迁移.有意思的是,“一步反应”的过渡态ATS(3/7)的结构是一个七元环过渡态,能垒为64.3 kJ/mol.此外,对ATS(3/7)进行了NBO 分析,表明N2 的H 原子电荷为0.490 e,而N1 的H 原子电荷为0.395 e,即N2的H原子比N1的H原子具有更强的亲电性,进一步证实N2的H原子比N1的H原子更易于迁移到配体CF3COO-带负电荷的O原子上.

Fig.3 Calculated Gibbs energy profiles for N2—H activation along mechanism A
接下来发生的是CO 插入.中间体A7 与一分子的乙腈(MeCN)配体配位得到更稳定的中间体A7MeCN,随后脱去TFAH和MeCN配体后生成中间体A8,接着CO与Pd原子配位形成中间体A9.尽管进行了多次尝试,还是未能找到TFAH或MeCN配体配位时CO与Pd原子配位的过渡态,分析原因主要可能是配体的空间位阻及较弱的供电子能力不利于CO与Pd金属中心配位.随后,探究了两种CO插入的可能方式:CO插入Pd—N1键或Pd—O键.此外,考虑到配体效应对反应的影响,也讨论了分别以溶剂MeCN,TFAH作为配体以及无配体的3种情况.计算结果表明,在羰基插入步骤中,MeCN作为配体无论在动力学还是热力学上都更为有利(图S2 和图S3,见本文支持信息).如图4 所示,相比于中间体A9,在羰基插入Pd—N1 键的过渡态N-ATS(10/11)MeCN中Pd—N1 键断裂,C—N1 成键生成中间体N-A11MeCN,能垒为103.9 kJ/mol;在CO 插入Pd—O 键中,通过过渡态O-ATS(10/11)MeCNPd—O 键断裂,C—O成键,能垒为106.4 kJ/mol,得到六元环中间体O-A11MeCN.

Fig.4 Calculated Gibbs energy profiles for CO insertion along mechanism A
基于两种CO插入方式所产生的六元金属环中间体N-A11MeCN和O-A11MeCN,在还原消除步骤中对应着两种可能的反应路径即C—O成键和C—N1成键.同样,也考虑了分别以溶剂MeCN,TFAH 作为配体以及无配体的3 种情况,结果表明,在还原消除步骤中,MeCN 作为配体也是优先发生的(图S4 和图S5,见本文支持信息).如图5所示,对比C—O成键和C—N1成键两种还原消除路径,可见,相对于中间体N-A11MeCN,C—O 成键还原消除的[N-A11MeCN→N-ATS(11/12)MeCN→N-A12MeCN]能垒比C—N1 成键的还原消除[O-A11MeCN→O-ATS(11/12)MeCN→O-A12MeCN]的低33.1 kJ/mol.因此,C—O成键的还原消除路径在动力学上要优于C—N1成键的还原消除路径.此外,对双MeCN配体的还原消除路径也进行了计算,结果显示,单MeCN 配体的还原消除路径在动力学上要优于双MeCN 配体的还原消除路径(图S6,见本文支持信息).最后在氧化剂CuO和TFAH作用下再生出Pd(TFA)2催化剂,完成整个催化循环.
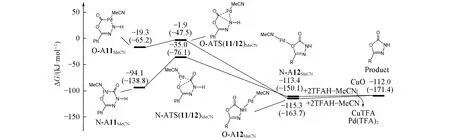
Fig.5 Calculated Gibbs energy profiles for reductive elimination along mechanism A
基于实验猜测的机理A(N2—H活化/CO插入/还原消除),计算结果表明,整个催化循环需要经历一个较高的过渡态ATS(8/9),能垒为134.2 kJ/mol(相对于中间体1a).因此,实验上提出的反应机理A能量上是不利的,可能存在新的反应机制/路径来阐明实验反应.
2.1.2 机理B(CO插入/N2—H活化/还原消除)不同于机理A(N2—H活化/CO插入/还原消除)能量上的不可行,在新提出的机理中,在N1—H活化得到中间体3后,先发生CO插入反应,再发生N2—H活化和还原消除.从中间体3开始,CO插入有两种可能的路径:CO插入Pd—N1键或Pd—O键(图6).在CO插入Pd—N1键的路径中,首先是CO中的C原子进攻中间体3的Pd原子中心,导致Pd与CF3COO-中的一个O断键,与CO中的C配位,得到稳定的中间体N-B4,该化合物中Pd(Ⅱ)原子配位饱和并满足18电子规则.随后CO 插入到Pd—N1 键,过渡态N-BTS(4/5)振动方式显示了Pd—N1 键的断裂和C—N1键的生成.这一过程(3→N-B5)需要克服102.0 kJ/mol的能垒,放热78.0 kJ/mol.CO插入Pd—O键的路径中,CO配位到中心Pd原子上得到稳定的中间体O-B4.随后CO插入Pd—O键,形成C—O键,该过程(3→O-B5)吸热4.2 kJ/mol,能垒为87.6 kJ/mol.中间体N-B5 和O-B5 发生N2—H 活化,同机理A 中的N2—H活化类似存在3种可能:三步机理、两步机理和一步机理(图S7和图S8,见本文支持信息).计算结果表明,一步机理所需克服的活化能垒最低,要优于前两种机理.通过NBO 分析中间体N-B5 和O-B5,证实它们的N1原子(-0.542和-0.622 e)比N2原子(-0.475和-0.360 e)带有更多的负电荷,不利于N1原子上的H原子离去,因此N2原子上的H原子更易发生一步机理直接活化.如图6所示,在一步机理中,中间体N-B5 要比中间体O-B5 稳定,相对于中间体N-B5,路径N-B5→N-TS(B5/A11)→N-A11MeCN的能垒较低为69.3 kJ/mol,而路径O-B5→O-TS(B5/A11)→O-A11MeCN的能垒较高为142.1 kJ/mol.因此,前者在热力学和动力学上都优于后者.后面的还原消除步骤与机理A的相同.

Fig.6 Calculated Gibbs energy profiles for CO insertion and N2—H activation along mechanism B
综上,Pd 催化氧化N—H 键羰基化反应合成1,3,4-噁二唑-2-酮杂环化合物新路径(机理B)的计算结果表明,此路径在能量上是更有利的.结合图3,图5和图6,可见,钯催化氧化N—H键羰基化反应合成1,3,4-噁二唑-2(3H)-酮杂环化合物反应的催化机理包含N1—H活化、CO插入、N2—H活化和还原消除(Scheme 4).其中在CO 插入步骤(N-B4→N-B5)是反应的决速步骤,该机理的活化能垒为102.0 kJ/mol.

Scheme 4 Proposed mechanism B for the Pd-catalyzed carbonylation
为了进一步分析机理A和机理B的竞争性,对决速步骤的过渡态进行结构分析.如图7所示,机理A中的决速步骤CO配位的过渡态ATS(8/9)中,Pd—CCO键键长0.303 nm,且Pd(II)金属中心配位不饱和.而机理B 中,CO 配位的过渡态N-BTS(3/4)中,Pd(II)中心原子分别与底物中的O 原子、N1 原子,配体CF3COO-中的O 原子和CO 中的C 原子配位形成一个稳定的平面四配位化合物,Pd—CCO键键长0.244 nm.所以其相应的过渡态N-BTS(3/4)(-2.8 kJ/mol)的相对能量要比机理A 中CO 配位的过渡态ATS(8/9)(47.6 kJ/mol)稳定.正是这种稳定的结构有利于接下来CO插入基元反应的发生,因此机理B中决速步骤经历的过渡态N-BTS(4/5)(-2.9 kJ/mol)也很稳定.

Fig.7 Optimized geometric structures of transition states ATS(8/9),N-BTS(3/4)and N-BTS(4/5)
2.2 取代基效应
根据Jiang等[16]的实验结果,对一系列苯甲酰肼底物在决速步骤羰基插入中的表现进行研究,包括供电子基团甲氧基(MeO)、叔丁基(t-Butyl)、苯基(Ph)和吸电子基团[氟(F)和硝基(NO2)](表1).

Table 1 Calculated the barriers for the rate-determining step and reaction energies of different substituents*
当底物取代基R为MeO,t-Butyl和Ph时,决速步骤的能垒分别为101.0,102.3和100.0 kJ/mol;当R 为F 和NO2时,决速步骤的能垒分别为101.1 和103.2 kJ/mol.可见,不同取代基对生成1,3,4-噁二唑-2(3H)-酮杂环化合物的活化能垒影响非常小.因此,钯催化羰基化反应合成1,3,4-噁二唑-2(3H)-酮杂环化合物对含有不同取代基的苯甲酰肼底物具有普适性,与实验结果一致.
3 结 论
采用密度泛函理论M06方法对钯催化苯甲酰肼的羰基化合成1,3,4-噁二唑-2(3H)-酮的反应机理进行了研究.基于计算结果,提出了一个新的反应机理,该反应的催化循环包含N1—H 活化、CO 插入、N2—H活化和还原消除4个阶段.首先通过CMD机理经过经典的六元环过渡态完成N1—H键的活化生成五元金属环中间体.从五元金属环中间体开始,存在两种机理:N2—H活化/CO插入/还原消除(机理A)或CO插入/N2—H活化/还原消除(机理B).计算结果表明,机理B不论在动力学还是热力学上均比机理A更为有利.在机理B中,先羰基插入Pd—N1键生成六元金属环中间体,接着通过一步机理直接活化N2—H键使H原子迁移到配体CF3COO-上,随后TFAH离去,接下来发生MeCN与金属中心配位还原消除,最终得到产物1,3,4-噁二唑-2(3H)-酮.CO插入Pd—N1键是整个催化循环的决速步骤,能垒为102.0 kJ/mol.此外,MeCN在催化体系中不仅是反应的溶剂,而且在还原消除步骤中以配体参与反应,能够有效降低反应所需的活化能.取代基效应的研究表明,钯催化苯甲酰肼的羰基化反应对于不同取代基取代的苯甲酰肼底物具有普适性.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210067.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中国药学药品知识仓库(2022年10期)2022-05-29
大学化学(2021年8期)2021-09-26
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
汕头大学学报(自然科学版)(2020年4期)2020-12-14
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
股市动态分析(2015年12期)2015-09-10
