
手性化合物立体结构鉴定中的若干关键共性科学问题探讨
2021-06-29 06:00王煦鑫朱华结
高等学校化学学报 2021年6期
王煦鑫,朱华结
(河北大学药学院,保定 070000)
随着天然有机化学、不对称合成化学以及相关手性材料学科的飞速发展,手性化合物的表征及立体结构的鉴定已经成为手性科学发展的关键科学问题.相关计算软件已经普及并被广泛使用,手性化合物立体结构的鉴定日益成为手性科技工作者的重要研究内容[1~10].但也出现了一些问题:(1)计算化学软件的使用,认为会使用软件就可以开展手性化合物相关性质的计算是一个误区;(2)计算中模型的简化,对于复杂且带有较长碳链的手性分子常要进行分子结构简化,但是简化模型使用的原因及如何简化具有科学性;(3)因不清楚相关方法的应用范围和前提条件而导致计算结论错误.为此,在计算能力得到极大提高的今天,依然有必要讨论这些常见的、具有共性的若干关键科学问题,从而提高我国手性化学学科的发展水平和质量.
1 软件问题
目前,在手性立体化学结构鉴定中,相关的计算软件比较丰富,主要分为构象搜索软件和模拟计算相关手性分子性质的软件两类.典型的构象搜索软件中有Sparton,Conflex(Barista),ComputeVOA,HyperChem,高斯新版本的Gaussian 16和其它软件.构象搜索软件的主要功能是可以对一个已知的结构使用不同的分子力场,如MMFF94S或其它力场进行构象搜素,记录每个在设定范围内的能量和该构象的坐标,并最终保存该设定范围之内的所有构象,用于后续的计算.
计算所选用的力场是一个关键问题,不同的力场得到的分子能量会有所差异.如果分子结构比较简单,如2-丁醇,无论采用何种力场计算,都能得到一致的稳定低能量构象.但手性分子结构通常比2-丁醇复杂得多,而且分子中的环系结构也比较复杂,会出现局部刚性很大的环结构.因此,在构象分析过程中必然导致3种主要结果:(1)不同起始结构的分子得到的稳定构象不同,这种情况在含有多个—OH结构的分子中更易出现;(2)最稳定的(能量最低)和较为稳定的构象结构未被找到,导致计算结果错误;(3)不同力场计算得到的结果在分子力场水平上出现较大差异。在实际计算工作中还会出现其它问题.
第一种和第三种结果比较容易理解,现对第二种结果举例以便于理解.如,采用Mosher酯方法鉴定分子1[11](图1)的—OH上C原子的立体结构.但对于手性中心旁边具有很大基团的手性分子,采用Mosher 酯方法可能出现错误的结论.如果要准确测定其立体结构,在(R)-1 的(R)-1a 和(R)-1b 2 个构象中,只有(R)-1a具有优势构象(即低能量),而在(S)-1的Mosher酯中,也需要(S)-1a处于优势构象才能保证鉴定结果正确.因此,对这个手性分子进行结构鉴定时,计算其不同构象的Mosher酯能量十分重要.文献[12]使用2种不同的软件(HyperChem和Barista)和2种力场(Amber和MMFF94)在完成系列化合物的构象搜索后,于近2万个的构象中才找到最低能量构象.该过程中,分子力场计算的能量窗口控制在0~25 kJ/mol.在B3LYP/6-31G(d)基组上完成该窗口内所有构象的计算后,所选择的构象能量窗口为0~12.6 kJ/mol.在排除掉能量简并的构象后,采用6-311+G(d)基组再次对所有的能量在0~6.3 kJ/mol范围内的构象进行优化计算.
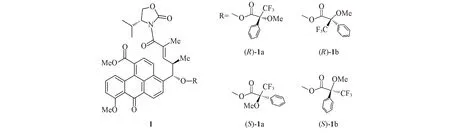
Fig.1 Four kinds of Mosher ester conformers of compound 1
两种不同的计算方法得到了相同的结果.对(R)-Mosher酯而言,能量最低的构象在(R)-Mosher酯中较多,即(R)-1b;对(S)-Mosher酯而言,能量最低的构象在(R)-Mosher酯中较多,即(R)-1a,但分布比例不同.由在B3LYP/6-31G(d)基组上计算所得比例可知,(R)-1a 的摩尔分数为30%,(R)-1b 为70%,二者比例约为1∶2(表1).在6-311+G(d)基组上计算所得比例约为1∶1.17.在B3LYP/6-31G(d)基组上进行(S)-1a 和(S)-1b 的能量分析时,得到的最稳定构象是(S)-1a,摩尔分数为54%;而在6-311+G(d)基组上的最稳定构象为(S)-1b,摩尔分数为58%.结果表明,在6-31G(d)和6-311+G(d)基组上计算的结果并不相同,但在6-311+G(d)基组上计算的结果比较可靠.根据Mosher酯进行绝对构型鉴定的可靠要求,在(R)-Mosher 酯中,最稳定构象是(R)-1a;在(S)-Mosher 酯中,最稳定构象是(S)-1a.而计算结果表明,实际最稳定构象分别是(R)-1b和(S)-1b.这与Mosher酯所要求的构象结论完全相反.因此,最后的实验结论肯定有误.
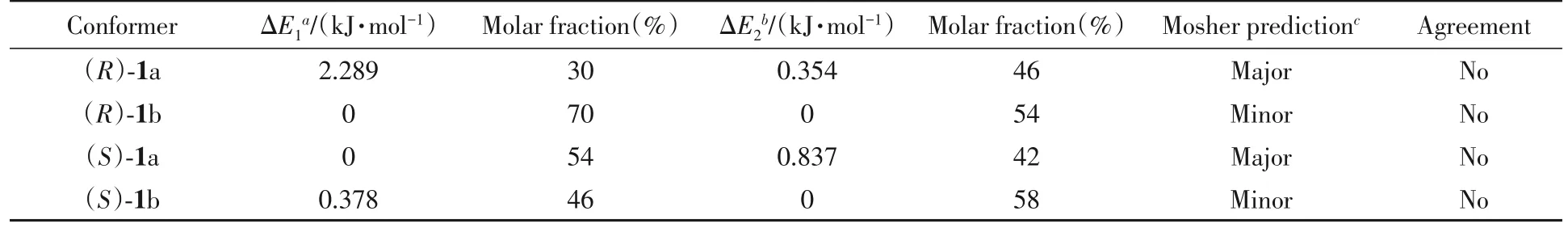
Table 1 Energies and distributions of the most stable and second stable conformers
无论是解释Mosher酯结论的可靠性还是计算分子的手性光谱等,准确得到分子的稳定构象是最关键的第一步.如,在化合物2(图2)中,由于存在围绕C1—C1′旋转产生的2个异构体,导致分子中同时存在2种手性,即轴手性(由于围绕C1—C1′旋转产生的2个手性异构体的旋转能量低,因此二者不能分离)和侧链分子中氨基酸C原子的手性[13].在1H NMR和13C NMR光谱中,出现了2套波谱.图3示出了这些化合物的1H NMR 光谱差异.这是由2 个不同构象,即(Sa,S)-2a 和(Ra,S)-2b 引起的构象异构.其中,确定优势构象的能量则依赖于量子化学计算结果.
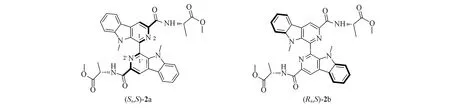
Fig.2 Two stable conformers of compound 2

Fig.3 1H NMR(A)and 13C NMR(B)spectra of compound 2
在计算过程中,若使用HperChem软件,可以先不定义二面角N2—C1—C1′—N2′为变量[14],先计算(Sa,S)-2a或(Ra,S)-2b的稳定构象.若使用Conflex(Barista)软件,则没有这种选择,在构象分析与计算时,同时得到(Sa,S)-2a与(Ra,S)-2b的构象并保存在同一个输出文件中.在后期的构象分析中,需要手动将构象(Sa,S)-2a与(Ra,S)-2b区分开,并分别用于后期的数据处理.在新版的Gaussian 16软件中也有二面角的选项.因此,使用HyperChem 或者Gaussian 16 软件均可方便地分析得到(Sa,S)-2a 和(Ra,S)-2b的构象.
该分子中有2个不同类型的手性中心,即轴手性和侧链氨基酸的手性.其中,经理论计算得到起主要作用的结果列于图4.二者的电子圆二色光谱(ECD)几乎对称,表明轴手性对ECD光谱的结构影响大,其侧链中的C=O以及共轭的CONH结构则对ECD的谱带几乎没有影响.
图4结果表明,确定绝对构型时,即使在生色团附近的手性中心(化合物2中与酰胺相连的C原子)也会因其它原因(化合物2中为轴手性)在ECD光谱结构中无法体现.因此,在实际工作中需要特别注意:(1)在含轴手性结构中,仅通过ECD不易鉴定其它位置的手性构型;(2)在不含轴手性的分子中,要尽可能对若干不同的可能结构分别进行计算.
通过对化合物2的构象分析比较发现,(Sa,S)-2a和(Ra,S)-2b的摩尔比经理论计算为4.8∶1,实验值为2.3∶1,所有计算均在B3LYP/6-311+G(d,p)上完成,溶剂中计算选用极化连续介质模型(PCM)[13].

Fig.4 Effect of axial chirality on ECD spectra of compound 2
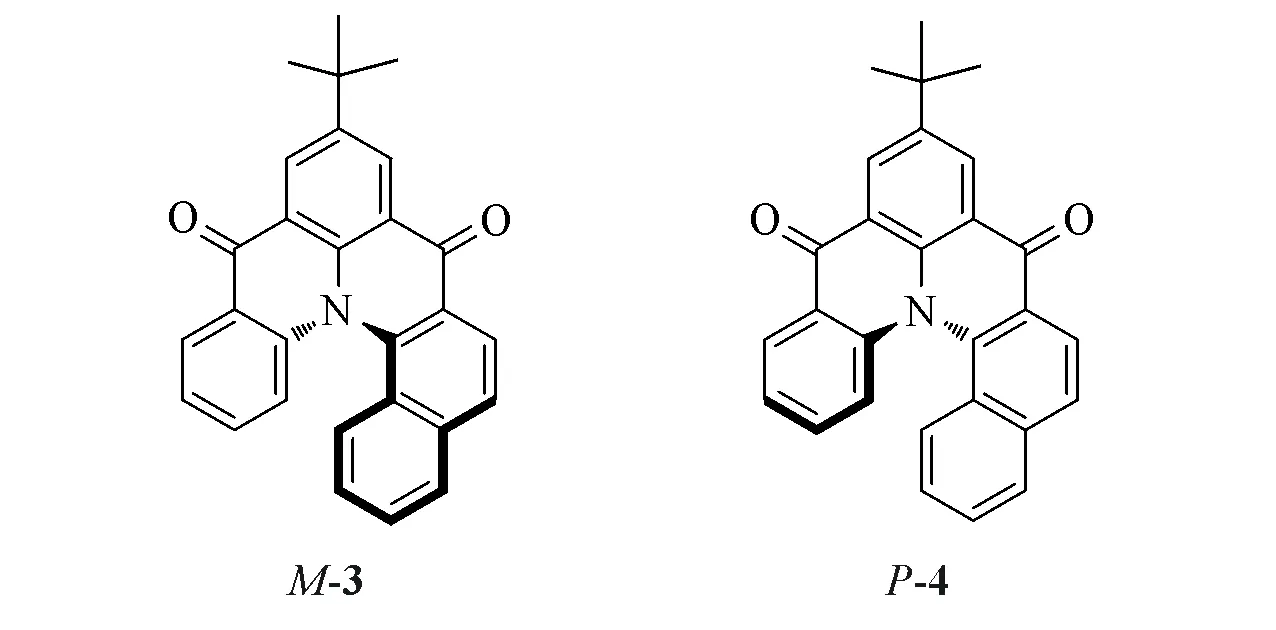
Fig.5 Enantiomeric structures of enantiomer compounds M-3 and P-4
在实际工作中,确定最稳定构象的分子十分重要,但有时仅靠软件还不够.据文献[15]报道化合物M-3比化合物P-4在能量上低0.0819 kJ/mol(图5).由于二者是对映体,能量相同,因此该结论肯定有误.错误的主要原因是没有准确分析t-Bu中3个甲基的相对位置.t-Bu在M,P构型的分子中应该分别具有2 个稳定的构象.对于M-3,经B3LYP/6-31G(d)基组计算得出构象M-3a 比构象M-3b 的能量低0.0819 kJ/mol.将构象M-3a与P-4a同时用于计算时,2个对映体的能量完全相同(图6).由于计算导致的能量误差小于10-6kJ/mol,可以完全忽略.因此,这里构象分析的关键点是t-Bu的空间构象.
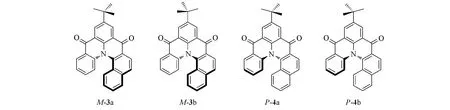
Fig.6 Four conformers of enantiomers 3 and 4
对映体过量百分率(e.e.,%)在生命起源中的影响较为突出.在M-3 和P-4 中,如果二者能量差0.0819 kJ/mol(表2),将导致M-3与P-4相差1.6%的e.e.值.后期的生命进化中,多余的M-3可能会以催化剂的形式参与各种反应,从而加速生命起源.在生命起源研究中,这个差异十分巨大,但在手性光谱计算模拟中不会有显著影响.因此,可靠而正确的构象分析在计算中十分重要[16].

Table 2 Calculated relative energy of the four enantiomers
2 计算中简化模型的使用问题
对于复杂且带有较长碳链的手性分子通常需要简化分子结构,简化模型使用的原因及简化方法值得研究[17].具有较长侧链的手性分子通常会把长侧链简化为短侧链,分别用于计算其电子圆二色光谱(ECD)、振动圆二色光谱(VCD)和旋光光谱(OR)等.其中的关键是,如果进行合适的简化,根据理论基础得到的理论结果能很好地代表其原来的手性分子结构的物理性质.
非手性分子5理论上存在4个稳定构象5a~5d(图7)[3].由于C1和C4之间的排斥力,构象5b能量很高,实际分布很少,可以认为不存在.构象5a和5b都具有对称面,因此其旋光值均为零.但构象5c和5d没有对称元素,因此这2个构象一定有旋光.构象5c在B3LYP/6-31G(d)//B3LYP/6-31G(d)基组上计算所得旋光值约为-2.61°,构象5d则为+2.58°.由于二者为对映体结构,因此,二者能量几乎相同,二者的旋光值之和几乎为零.

Fig.7 Four conformer structures of compound 5
手性结构6 也有4 个稳定构象6a~6d(图8)[3].在B3LYP/6-31G(d)//B3LYP/6-31G(d)基组上计算得到的3个稳定构象的旋光值和能量列于表3.
图9示出了化合物6a,6c和6d结构的纽曼投影式.可见,构象6c与6d的旋光符号相反,旋光值比较接近,经Boltzmann 统计加和后二者对旋光的净贡献仅为-1.7.因此,构象6c和6d的旋光贡献可以忽略不计,主要的旋光贡献来自最稳定的构象6a.

Fig.8 Four stable conformers of compound 6

Table 3 Calculated relative energies and optical rotation values of conformers 6a,6c and 6d

Fig.9 Newman structures of chiral 2-Cl butane and their relative energies and specific optical rotation values
随着碳链的增加,类似6c和6d的构象结构会有很多[18].但这些构象对对旋光的净贡献不大.
构象对的存在致使所有构象对的旋光值对整体旋光值影响不大,这是使用简化模型的理论基础.考虑到计算的误差,使用简化模型计算在节约计算时间上占更大的优势.在构象搜索中,由于软件本身的算法和计算人员对参数的设置等问题,一些较低能量构象不易被发现,会造成计算失误.因此,使用简化模型有时能获得更可靠的结果.
矩阵模型理论计算结果表明,在距离手性碳最远到第3个碳时,其质量对旋光的影响已经很小[19].该结论与构象对紧密联系,因此,可以使用简化模型进行计算.
此结论虽然是从分析分子的旋光得到的,但在实际中可以广泛应用于其它类型的计算.侧链分子结构不仅局限于饱和的碳原子,同样可以用于ECD 和VCD 计算中应用的模型分子.VCD 技术是近年来兴起的手性化合物鉴定方法,具有良好的应用前景[20,21].其特点是IR与VCD信号同步测定,并合成在一个图片中,分别研究在IR信号中不同键的振动时,所对应的VCD信号的变化.例如,从Peperomiaobtusifolia中分离得到的新化合物Chromanes(7和8)[22,23],在VCD研究中使用模型分子9和10[24],将其绝对构型纠正为R构型.将α,β-丁二烯类和异戊二烯分别计算简化为乙烯和乙基基团用于计算,图10给出理论计算所得分子(+)-9的VCD光谱、分子7的实验VCD光谱其相关的IR光谱.
文献[25]未对天然产物分子11 的立体结构进行鉴定,但报道了其ECD 光谱.该结构中存在几个较大的取代基R,其位置与手性中心相差较大.基于单手性中心结构,可以将其简化为模型分子12或更简单的模型分子13来计算其OR或者ECD,从而确定其C13位的绝对构型.使用文献中默认的顺式结构,通过2种不同的简化模型讨论不同数量的生色团对ECD光谱的影响.
对化合物11的绝对构型进行分析时,如果使用模型12替代,可以得到2个稳定构象;如果使用模型分子13替代,则只有1个稳定构象.模型12和13均为刚性结构,在B3LYP/6-311+G(d)基组上使用模型(R)-12和(R)-13进行优化和ECD计算发现,结果有较大差异(图11).通过计算可以确定其立体结构.由图12可见,在360 nm处为正峰的异构体构型为(R)-11,在该处为负信号的立体结构为(S)-11.因此,选对合适的模型在ECD的计算中可以达到事半功倍的效果.

Fig.10 Structures of compounds 7 and 8 and their simplified models 9 and 10,predicted VCD and IR spectra for compounds 7 and 9
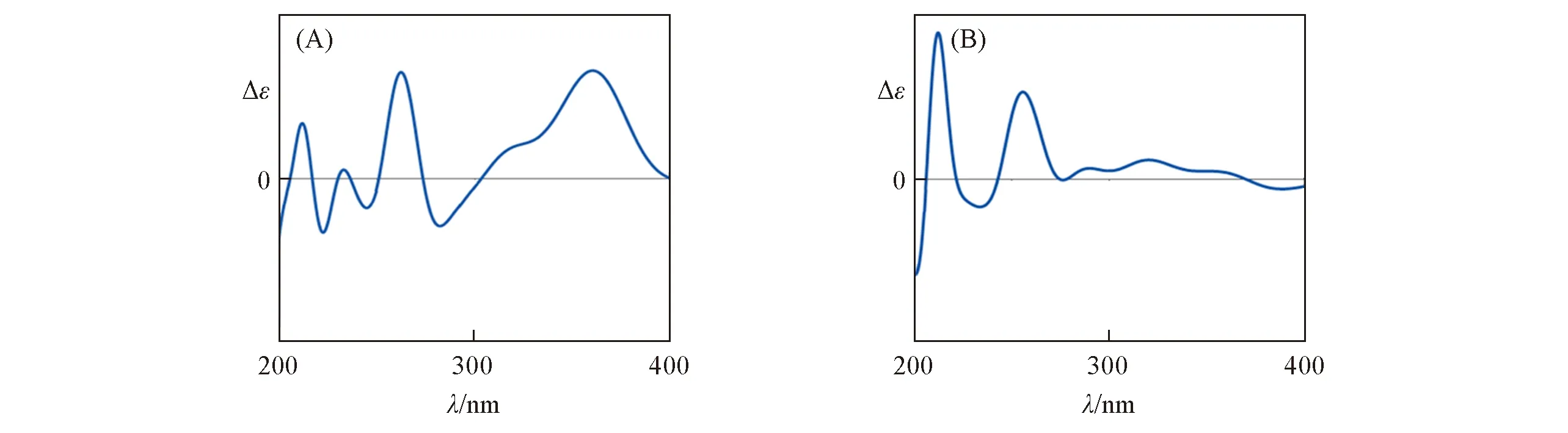
Fig.11 ECD spectra of model molecules(R)-12(A)and(R)-13(B)
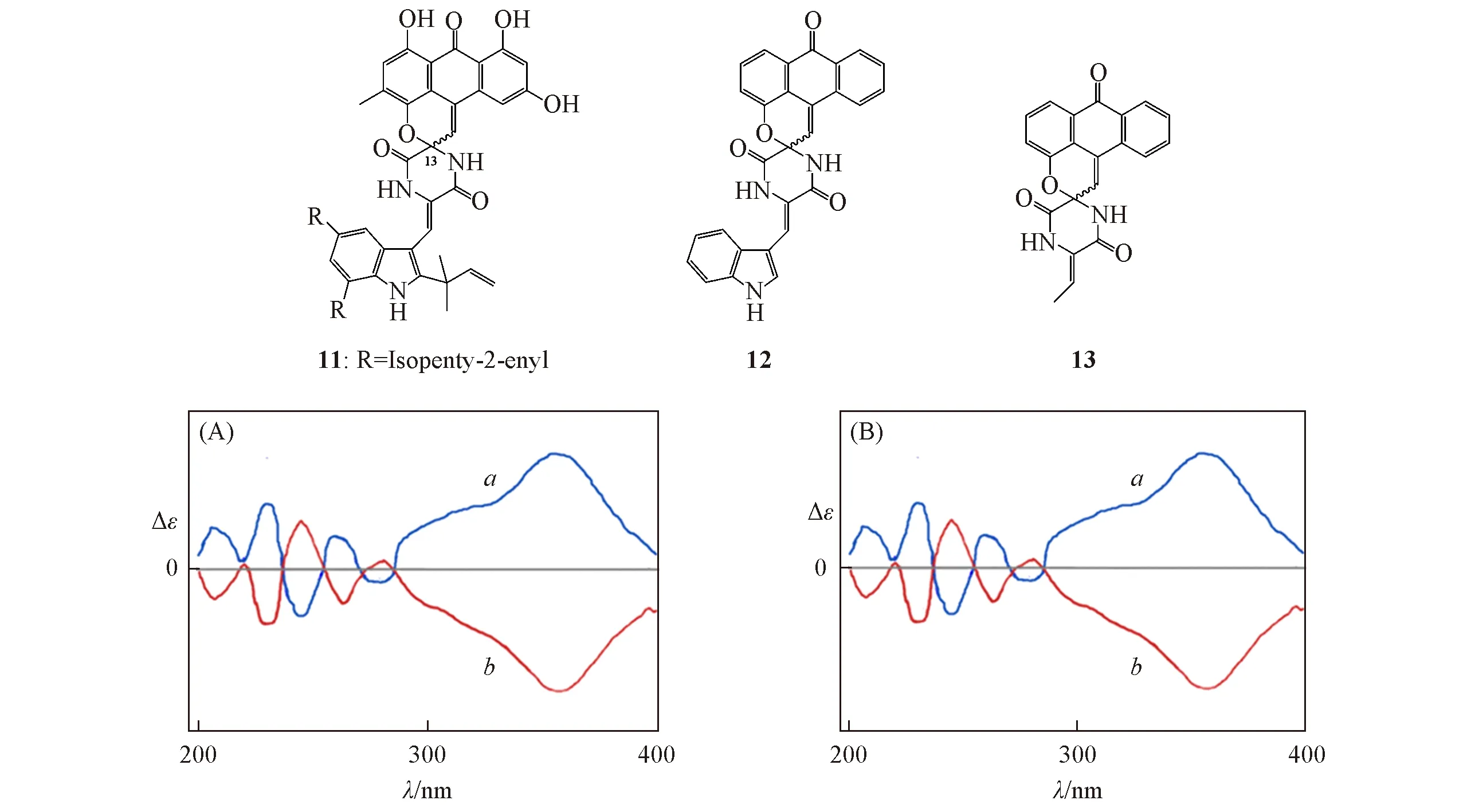
Fig.12 Compound 11 and its simplified models 12 and 13,and their ECD spectras for the isolated enantiomers
对于类似化合物11的结构,若对全部分子进行构象分析,其稳定构象至少有20个.而如果使用模型12来计算,其稳定构象只有2个,仅就构象数目而言,计算数量减少90%以上,考虑到原始分子11比模型12至少多15个重原子,其单一构象计算所需要的时间比单一模型12的构象也要多.因此,实际上计算量远远大于10倍.
从计算的精度来看,利用模型12 能很好地模拟原始分子11 的ECD 光谱结构,表明简化成功.而模型分子13因为少了1个吲哚生色团,导致360 nm附近的(+)-Cotton信号几乎消失,影响了最终的构型判断.这表明合适的模型选择对于准确判断分子的绝对构型十分重要.
在旋光计算或者VCD计算中,如果手性分子中存在长链结构,且距离手性中心较远,通常可以简化为含1~3个碳原子的1个取代基.在ECD 计算中,如果侧链上有生色团,可以根据生色团的位置进行相应的简化;如果没有生色团,则可以直接简化为1~3个碳原子的取代基.
3 计算方法的应用范围和前提条件
不清楚相关方法的应用范围和前提条件将导致在计算过程中出现错误.通常情况下,13C NMR的计算没有限制条件,适合所有类型的手性分子立体结构的鉴定,最终确定手性分子的相对构型结构.如,在天然产物14的结构改造中[Scheme 1(A)],使用乙酰氯得到产物15和16,其13C NMR 与原料14的相差很大.而原料14 与氯乙酰氯反应[Scheme 1(B)]所得产物的13C NMR 谱与原料14 的十分接近,得到正常的酯化产物[26].

Scheme 1 Synthetic routes of compounds 15 and 16(A)and normal product 17(B)
对化合物14进行结构改造,分别得到产物15,16和17,产物15和16的结构变化最大.原结构中的环氧乙烷在开环后与C2进一步反应,生成新的环系结构,化合物15有一个新的峰δC95.24(化合物16:δC95.95),而原来的δC79.03处C2峰消失.δC95.24处的峰归属为张力较大的环上的C原子,即新环上的C2.为了进一步确证化合物15 的结构,对其进行了13C NMR 计算.用Barista 软件在MMFF94S分子力场下进行构象搜索,将所得的低能量构象在B3LYP/6-311+G(d)基组下进行优化和13C NMR 计算,将计算的碳谱与实际测定的碳谱进行对比(图13).结果表明,实验与计算的C2化学位移差值仅为1.82,在误差范围内.亚甲基C13位移值δC73.64与计算得到的位移值δC79.77相差6.13,属于正常误差范围内.因此,化合物15中环氧丙烷环结构是合理的.
由于ECD中手性中心与生色团的位置密切相关,因此,在很多绝对构型鉴定中,分子中饱和结构环系分子的绝对构型完全取决于其同位素光谱确定的相对构型.如果相对构型错误,其ECD光谱并不能反映出来,常常导致绝对构型错误.例如,化合物18的生色团非常少[27](图14),其相对构型虽然可通过二维NMR光谱确立,但是需要X射线衍射实验进一步确证.因此,通过ECD鉴定其绝对构型是可靠的.相似的例子很多,尤其在天然产物结构的绝对构型研究中频繁出现,需要高度注意.
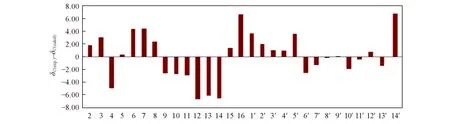
Fig.13 Relative errors between the calculated 13C NMR of compound 15 and its experimental data[δC(exp.)-δC(calcd])
计算ECD 光谱时尤其要注意手性中心与生色团间的相对位置.通常,如果手性中心距离生色团的位置超过2 个sp3杂化C 原子或者其它原子,其生色团对其手性中心的ECD 光谱的影响将会很小.因此,距离生色团在2 个原子之内的手性中心的立体结构鉴定较为可靠.如化合物19[28](图14)的手性中心距离—CHO 或者C=C 键这2 个较弱的生色团都较远,故几乎不能用ECD 来鉴定,但使用OR,ORD 或者VCD 方法可以鉴定其绝对构型.在绝对构型研究中,利用简化模型时一定要注意不能把在手性中心附近的生色团大量省略,否则极易造成计算错误,从而导致最终鉴定错误.
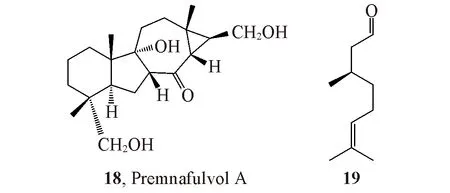
Fig.14 Structures of compounds 18 and 19
4 总结与展望
手性分子的旋光(OR)及VCD与其分子中远离手性中心的构象分析关系密切,而可靠的构象分析十分关键.对于长的侧链取代基,如果不存在手性中心,可以简化为1~3个碳的取代基结构,以简化计算.ECD与手性中心附近的生色团的位置和数量有关.在简化模型时,要注意保留必要的生色团结构和数量.在13C NMR 的计算中,手性构型的变化会导致手性中心附近碳原子化学位移发生变化,而手性中心的碳原子化学位移反而变化较小或者不变化.计算过程中,柔性链构象的变化对13C NMR影响较小.目前,手性计算软件发展速度快,应用范围日益广泛.只有充分认识手性分子的结构特点,了解所计算内容与结构的关系,并认识到相关软件的功能与不足,结合计算方法的应用范围和使用简化模型等,才能可靠地解决手性分子的立体化学问题.
猜你喜欢
分子催化(2022年1期)2022-11-02
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
北京航空航天大学学报(2017年10期)2017-04-20
温州大学学报(自然科学版)(2016年1期)2016-10-27
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
应用化工(2014年7期)2014-08-09
航天返回与遥感(2014年4期)2014-07-31
郑州大学学报(理学版)(2014年3期)2014-03-01
无机化学学报(2014年5期)2014-02-28
