
以言语缓慢起病损伤丘脑及脑干的肝豆状核变性1例报告并文献分析
2021-05-25 01:58:32熊泽辉陈晓露
中风与神经疾病杂志 2021年4期
熊泽辉,熊 涛,陈晓露,洪 周
肝豆状核变性,又称威尔逊病(Wilson disease,WD),是一种罕见的铜代谢异常,其特征是铜在肝脏、大脑和其他器官中的异常积聚。该病临床表现多样,以肝脏和神经系统异常临床表现为主要特征。肝型表现为肝细胞功能障碍,范围从脂肪变性到肝炎、纤维化及急性肝衰竭,神经型表现为神经精神症状,如震颤、说话困难、肌肉僵硬及性格变化[1]。该疾病由ATP7B中的纯合或复合杂合突变引起,在大多数人群中估计患病率为1/30000,在普通人群中突变载体频率为1/90[2]。因肝豆状核变性的临床变性具有异质性,目前仅以言语障碍起病的报道少见,容易误诊、漏诊,现结合相关文献报道以言语缓慢起病损伤丘脑及脑干的肝豆状核变性1例,并结合相关文献分析,为今后对该类疾病诊断和治疗提供借鉴。
1 病例资料
患者,女性,32岁,以“言语缓慢3 m余,加重1 m”于2020年9月1日于武汉协和医院入院。患者约3 m前出现言语缓慢,鼻音重,伴口角流涎,饮水、吃饭速度较前减慢,曾到当地市中心医院就诊,行头部磁共振平扫+增强扫描示:双侧大脑半球、基底节区、丘脑、脑干见对称性斑片状长T1、T2信号(见图1),FLAIR序列呈高信号;增强扫描未见明显强化(见图2)。骨髓活检:HE及PAS染色示送检骨髓增生较活跃(约60%~70%),粒红巨三系细胞增生,考虑“脱髓鞘脑病、中毒性脑病或代谢性脑病可能”,经激素治疗后,患者症状仍渐进性加重。遂于2020年9月1日,来武汉协和医院就诊。既往史、个人史无特殊;家族史:患者亲哥哥25岁以“行走不稳,伴双手抖动”起病,疑似肝豆状核变性,现已故。入院后查体: T:36.2 ℃,P:74次/分,R:16次/分,Bp:126/68 mmHg。一般内科查体未见异常。神经系统查体:神志清楚,言语迟缓,劲软,吐词欠清晰,可见舌肌震颤,余颅神经检查未见异常,四肢肌力Ⅴ级,四肢肌张力未见明显异常,共济运动正常,感觉系统检查未见异常,四肢腱反射(),病理征(-),Kerning征(-)。入院后完善相关辅助检查:血常规:白细胞1.59(3.5~9.5 G/L),红细胞3.47(3.8~5.1 T/L),血小板34 G/L(125~350 G/L);肝功能:总胆红素29.0(5.1~19.0 μmol/L),直接胆红素8.7(1.7~6.8 μmol/L),丙氨酸氨基转移酶47(5~35 U/L),天门冬氨酸氨基转移酶正常;凝血功能:D-二聚体 1.14(<0.5 mg/L FEU),PT 16.7(11.0~16.0 s),INR 1.38(0.80~1.31),APTT 39.1(28.0~43.5 s),FIB 1.23(2.0~4.0 g/L),血铜蓝蛋白:26(220~580 mg/L),Coomb’s试验阴性。肝胆脾胰彩超:肝形态失常,质地不均,门静脉增宽,脾大、脾静脉增宽。双眼裂隙灯下角膜缘可见棕绿色色素环(K-F环)。患者基因中检出 ATP7B 基因有2个突变:(1)c.2333G>T,p.R778L,为杂合错义突变;(2)c.2621C>T,p.A874V,为杂合错义突变;患者儿子基因中检出ATP7B 基因有1个突变:c.2621C>T,p.A874V,为杂合错义突变(见图3)。结合病史及辅助检查,诊断为肝豆状核变性,嘱患者限制铜的摄入,予以促进铜的排泄等治疗,并建议行脾脏切除。
2 讨 论
肝豆状核变性因ATP7B纯合或复合杂合突变(存在两种不同的突变等位基因)导致无铜蓝蛋白快速降解和胆汁铜排泄失败,铜在肝、脑、角膜、肾和其他组织中积累引起的海绵状变性、脱髓鞘及胶质增生[3],故临床上患者可出现外系症状,精神行为异常,肝炎及肝功能不全等[1],临床表现无特异性,极易漏诊误诊。本病例患者为女性,以“言语缓慢”为首发症状,曾于当地医院就诊,考虑“脱髓鞘病变、中毒性脑病或代谢性脑病可能”,经激素治疗后,患者症状渐进性加重,分析其诊断不明的原因有:(1)患者因“言语缓慢”就诊,可能的机制是因双侧基底节受损,导致神经递质传导障碍、运动神经元突触兴奋性增加,引起发音肌及协同肌功能障碍,导致发音动作、音量、速度失节律,本质上属于外系症状,目前文献报道甚少;(2)典型的肝豆状核变性患者的头部磁共振成像中常见的异常主要位于基底神经节(壳核和尾状核)[4,5],丘脑、中脑和脑桥少见。
WD患者可以是一种致病突变的纯合子,也可以携带两种不同的致病突变的杂合子。本例受检者中检出ATP7B基因有2个突变:(1)c.2333G>T,p.R778L,为杂合错义突变;(2)c.2621C>T,p.A874V为杂合错义突变。上述ATP7B 基因位点致病性的已在相关文献报道[6,7]。依据《EASL临床实践指南:肝豆状核变性》[8],该患者诊断为肝豆状核变性。关于肝豆状核变性的治疗,D-青霉胺是治疗肝豆状核变性首先的药物,该药物不仅络合血液及组织中过量游离铜从尿中排除,而且能与铜在肝中形成无毒的复合物而消除铜在游离状态下的毒性,考虑少数患者因青霉胺出现白细胞减少及骨髓抑制,目前患者存在白细胞、红细胞及血小板减少,故暂不使用青霉胺,予以口服甘草锌抑制铜在肠道的吸收。此外肝豆状核变性的患者一般应至少在药物治疗的第一年避免食用铜含量非常高的食物(例如贝类、坚果、巧克力、蘑菇和器官肉)[9]。除了特定的抗铜治疗外,对症治疗在治疗肝豆状核变性中也很重要,尤其是治疗肝损伤或肝功能衰竭的并发症[10]。Poujois等[11]人研究发现因肝豆状核变性导致肝硬化或急性肝衰竭的患者,肝移植可以延长患者的生存周期,此外肝移植也可改善肝豆状核变性患者的神经症状。目前相关机制尚不明确,可能在大脑中存在三磷酸腺苷酶(ATP7A和ATP7B,以ATP7A为主)转运蛋白,肝移植后ATP7A可能会补偿大脑中ATP7B的缺乏,因此大脑不会因为肝功能障碍而承受铜过载。本病例中患者出现肝硬化,脾大,白细胞、红细胞、血小板减少,脾脏切除及肝移植可作为考虑的治疗方案。
综上所述,肝豆状核变性临床表现复杂多样,鉴别诊断须从肝脏及神经系统两个方面主要征象考虑,尤其是以外系症状首发者,包括因口-下颌肌张力障碍引起的言语缓慢,需考虑本病的可能,同时需完善相关辅助检查进行诊断和鉴别诊断。另外由于肝豆状核变性会同时累及多个系统,对患者的病情须综合评估,制定适宜的治疗方案,才能使患者获益最大。
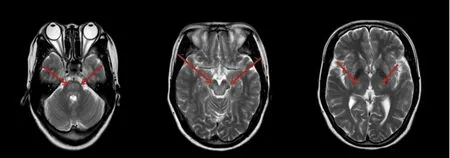
图1 头部磁共振T2序列:双侧丘脑、中脑、脑桥异常信号(箭头所示)

图2 头部磁共振增强:双侧丘脑、中脑、脑桥未见强化
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
现代临床医学(2021年4期)2021-07-31 07:56:08
种子(2021年3期)2021-04-12 01:42:22
中兽医学杂志(2019年1期)2019-01-06 23:48:45
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
罕少疾病杂志(2016年4期)2016-03-11 16:34:38
首都医科大学学报(2015年4期)2015-12-16 13:00:08
浙江医学(2014年17期)2014-04-13 10:13:16
