
以不自主抖动为首发表现的脊髓小脑性共济失调13型1病例报告
2021-05-25 01:58:30李旭颖王宪玲
中风与神经疾病杂志 2021年4期
郝 静,李旭颖,王宪玲
常染色体显性遗传性小脑性共济失调(ADCA)也称为脊髓小脑性共济失调(Spinocerebellar ataxia type,SCA),是一种起病隐匿,缓慢加重的神经系统变性病。其临床表现非常广泛,包括小脑性共济失调、眼动障碍、锥体外系运动障碍、视神经萎缩、视网膜病变、认知障碍、周围神经病及癫痫等[1]。由于各亚型间存在着巨大的重叠,临床上对于特定亚型的诊断非常困难。随着基因检测技术的发展与应用,目前已鉴定出40余种亚型。SCA13型是临床上罕见的一种类型,由于缺乏足够的临床数据,SCA13型的发病率尚不清楚。SCA13型在中国人群中罕见[2],此前国内仅报道过1个家系[3],该家系中发现了KCNC3基因未被文献报道的新的突变位点c.1018G>A。现将我们发现的另1例SCA13型的病例报道如下,在该病例中以不自主抖动为首发表现,既往尚未见过类似报道。
1 病例资料
患者,男性,17岁,慢性起病,主因“双手抖动10余年,言语不清、行走不稳7 y。”以“行走不稳待查”收入院。患者10余年前无明显诱因出现双手不自主抖动,表现为取物时抖动明显,安静时也可出现。双手活动欠灵活,可完成日常生活。无明显加重缓解因素。7 y前无明显诱因出现言语不清,语速减慢,说话断续。同时出现行走不稳,左右摇晃,无法走直线。未出现跌倒。偶有饮水呛咳。入院查体:血压:115/79 mmH,心率80次/分,呼吸20次/分,体温36.5 ℃。神清,言语缓慢,粗测高级皮质功能基本正常。双侧瞳孔等大正圆,直径约3.0 mm,对光反射灵敏。双侧眼球活动正常,双侧鼻唇沟对称,伸舌居中,咽反射存在,余颅神经查体未见明显异常。双手可见不自主抖动。四肢肌力5级,双下肢肌张力略增高。双下肢腱反射(),双侧胸大肌反射阳性,双侧hoffmann征(-),双侧巴氏征(-)。深浅感觉查体未见异常。双侧指鼻,跟膝胫试验欠稳准,轮替试验欠灵活,闭目难立征阴性。一字步不能。脑膜刺激征阴性。个人史:无难产窒息病史,生长发育史正常。家族史:家族中未有类似疾病史。
辅助检查:头部磁共振成像(MRI)(见图1):双侧小脑半球脑沟増宽、加深,小脑萎缩,左侧颞角扩大。核磁共振波谱图(MRS):右侧小脑感兴趣区NAA浓度(NAA/Cr:1.04),CHo浓度(CHo/Cr:0.97);左侧小脑感兴趣区NAA浓度(NAA/Cr:0.94),CHo浓度(CHo/Cr:1.01)。眼科检查未见KF环。腰穿压力 175 mmH2O,脑脊液白细胞计数1×106/L,脑脊液生化:脑脊液蛋白13 mg/dl,涂片找抗酸杆菌、细菌、真菌、墨汁染色、TORCH病毒检测、24 h CSF IgG鞘内合成率及寡克隆带均未见异常,自身免疫性脑炎、副肿瘤抗体:阴性。MMSE(简易精神状态检查量表)评分:26分(学历:初中)。MOCA(蒙特利尔认知评估量表):20分。画钟实验1分。余常规化验未见异常。
在本例中,排除常见的CAG重复序列导致的SCA亚型后,对患者及其父亲进行全外显子测序(母亲拒绝检测)。分析结果提示,患者的KCNC3基因检测出杂合突变,父亲为野生型,但由于缺少患者母亲的基因信息,目前尚不能确定该基因突变是来源于母亲还是新发突变。在本例中,该基因发现一处已报道的致病错义突变c.1268G>A,导致第2号外显子(总共5个外显子)423位点上的精氨酸被组氨酸取代(R423H突变),且功能预测为临床有害。由于:(1)KCNC3基因是SCA13的唯一致病基因;(2)该突变已有文献报道,在群体中的携带频率极低,功能预测为临床有害突变,且遗传模式符合常染色体显性遗传,ACMG预测为致病变异(PAT,PM1+PM2+PP3+PP2+PP5);(3)功能研究结果提示:KCNC3编码电压门控钾离子通道蛋白3(Kv3.3)。该基因在脑中高表达,尤其在小脑,从遗传和机制上能够解释本病例。进行一代测序验证(见图2),结果证实了突变的真实存在,符合SCA13型。在治疗上主要给予B族维生素(维生素B1、维生素B6、甲钴胺)营养神经等治疗。半年后随访未进一步进展。
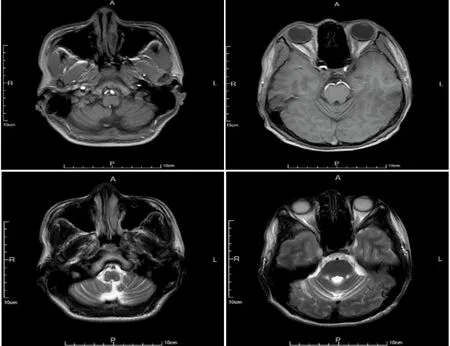
图1 患者头部MRI:可见双侧小脑半球脑沟増宽、加深
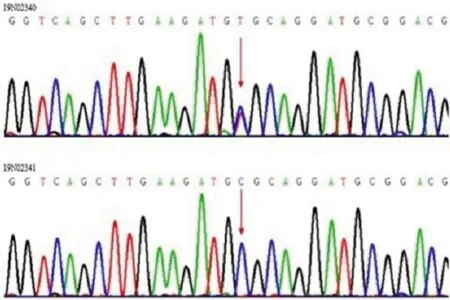
图2 患者(上)及其父(下)基因检测图谱
2 讨 论
SCA是遗传性共济失调类疾病的主要类型。一项纳入了1062篇文章的荟萃分析发现,在SCA的患者中非共济失调类的临床表型也很常见[4]。该患者首次以不自主抖动为首发症状,该临床表型尚未在SCA13型患者中进行过报道,容易造成在疾病早期误诊的可能。其次,该患者在安静状态及活动时均可见双手非对称性的抖动,接近物体时抖动无明显加重(未行震颤分析),不符合典型的小脑性震颤。结合患者的发病年龄,首先需要除外其他运动障碍性疾病。不自主抖动属于运动障碍的范畴,运动障碍是SCA重复扩增型患者的常见症状。高度提示在不同的亚型间临床表型存在着巨大重叠。头部MRI是SCA患者首选的影像学检查。随着疾病的进展,脊髓小脑性共济失调的影像学可表现为脊髓萎缩、橄榄桥小脑萎缩和皮质小脑萎缩。在该患者中,可观察到明显的双侧小脑半球的萎缩,符合SCA的影像学表现。
相对于常见的SCA亚型,SCA13型的发病机制研究较少,基因KCNC3(也称为Kv3.3)是SCA13型的唯一致病基因,位于染色体19q13.3-19q13.4,编码Kv3.3电压门控钾通道。该通道主要分布在小脑浦肯野细胞、深部神经元、颗粒细胞等[5]。KCNC3基因的突变可能会导致Kv3.3通道蛋白功能改变,影响小脑的抗氧化应激的能力。该基因突变已在非洲爪蟾卵母细胞中得到验证[6],SCA13型突变体主要影响了小脑神经元兴奋性以及KCNC3表达的其他区域的神经元功能。目前在斑马鱼[7]、小鼠模型[8]中也进行了相关突变研究。在R424H突变的小鼠模型中,突变的结果会导致钙通道的改变,影响神经元兴奋性并导致小脑神经元的死亡,因此在SCA13型的患者中可观察到明显小脑萎缩。
既往已有文献报道,证实R423H为致病性突变[9]。可有研究表明,SCA13型发病早晚和临床症状可能与Kv3.3的激活转移的方向有关[10]。R423H突变体可以抑制Kv3.3电流幅度,还可以显著改变通道门控功能[9]。F448L和R423H突变体对通道门控有相似的作用[11],F448L会在超极化方向上改变通道激活电压的依赖性,减慢通道关闭的速度。近年来,Duarri等人验证了一个新的突变体V535M以及两个可能的突变体D129N和S391G[10]。其中V535M和D129N也可以将Kv3.3通道激活转移到超极化电压。这4个突变体均改变了通道门控的功能,都表现为早期发病。R420H突变的患者临床上表现为成年起病。经过功能验证,R420H和R423H突变均位于Kv3.3通道S4跨膜区段中,均可产生显性负性的功能亚基,但是R420H并未导致Kv3.3门控功能发生改变[11]。因此,发病年龄的早晚在很大程度上取决于通道门控功能的改变。同时有研究发现Kv3.3通道激活的去极化可能与痉挛性共济失调步态有关[10]。此次报道的R423H突变的患者以锥体外系症状为首发表现。运动障碍考虑与基底节的神经元变性相关。震颤、肌张力障碍等运动障碍在SCA经典突变亚型中少见,该病例提示包括震颤在内的运动障碍似乎也是SCA13型临床谱系的一部分,扩大了SCA13型的临床表型库,同时也在一定程度上支持了基因型-临床表型的关联。
针对SCA的治疗,目前尚无特效的办法来阻止疾病的进程[1]。国外利用慢性丘脑刺激治疗了1例SCA2型患者的震颤[12]。在该患者中,在药物治疗方面主要给予了营养神经的B族维生素治疗,并未找到特效的疗法来延缓疾病发展。由于不同的亚型其发病机制存在一定的差异,已有试验针对特定的亚型得到了某些有意义的结果。例如服用乙酰唑胺改善了SCA6型患者的共济失调的症状[13]。目前有关SCA的研究还不够充分,但是新的技术如蛋白组学分析等可帮助更好的寻找药物对应的治疗靶点,从而为寻找新的治疗方向提供可能。
对于脊髓小脑性共济失调,目前研究有限,在临床上对于有共济失调症状的患者,一定要追问其家族史,必要时行基因检查。以锥体外系为首发表现的共济失调的患者,应该考虑到该病的可能。临床上需要多多留意相关患者,为进一步深入了解疾病提供帮助。
猜你喜欢
中国神经精神疾病杂志(2022年3期)2022-07-14 02:23:42
世界科学技术-中医药现代化(2021年8期)2021-12-21 07:04:52
中国临床解剖学杂志(2021年2期)2021-04-19 14:52:46
创新作文(小学版)(2019年4期)2019-07-24 09:03:42
哲思2.0(2017年12期)2017-03-13 17:45:04
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
中国医药科学(2015年24期)2016-03-07 15:32:46
西南军医(2016年2期)2016-01-23 02:14:10
山东医药(2015年40期)2015-02-28 14:28:45
