
副肿瘤性脑干脑炎合并吉兰-巴雷综合征1例报告并文献复习
2021-05-25 01:58付胜奇张洪涛张淑玲
中风与神经疾病杂志 2021年4期
张 津,宋 良,付胜奇,张洪涛,张淑玲
吉兰-巴雷综合征(Guillain-Barres syndrome,GBS)是一种自身免疫介导的周围神经病,主要损害多数脊神经根、周围神经及脑神经的疾病,然而关于此类患者是否伴随中枢神经系统肿瘤性病变,仍存在较大的争议。现将我院收治的1例吉兰-巴雷综合征合并副肿瘤相关的脑干脑炎报道如下,并对有关文献进行复习。
1 临床资料
患者,男性,57岁,因“右肺腺癌术后5 y,加重伴咳嗽咳痰半月余”于2019年6月23日入住我院放疗科。5 y前患者因右侧胸痛伴咳嗽、气促,就诊于当地人民医院,胸部CT示右肺上叶占位,遂行手术切除治疗,术后病理提示:右肺下叶中分化腺癌,右胸膜见腺癌浸润。术后多次行放、化疗(先后2周期TP,6周期培美曲塞,胸部放疗一疗程)及靶向药物(凯美纳125 mg tid)治疗。2018年12月1日入院复查颅内转移瘤,给于颅内转移灶伽马刀放射治疗,期间肺癌外周血基因检测EGFR19外显子突变。2019年2月14日服用“吉非替尼”至今。15 d(2019年6月10日)患者咳嗽咳痰较前明显,黄白痰、偶有痰中带血,伴恶心、间断呕吐胃内容物,无呕血、咖啡样物,无腹痛、发热等不适,随至我院住院治疗。入院第11天患者出现左下肢无力,不伴其他神经功能缺损症状,患者尚能自行如厕、走路。入院第12天患者左下肢无力逐渐加重,伴饮水呛咳、吞咽困难。入院第16天患者出现右下肢肢体无力,且左下肢无力活动不能,呈持续性,请神经内科会诊查体示:意识呈嗜睡状态,构音障碍,高级智能明显减退。双侧额纹变浅,闭目无力,双侧瞳孔等大等圆,直径约3.0 mm,光反射存在,双眼向左侧凝视麻痹,双鼻唇沟对称,伸舌左偏,咽反射消失,30 ml哇田饮水试验阳性,四肢肌张力减低,双上肢肌力4-级,左下肢肌力0级,右下肢肌力3级,双上肢腱反射(-),左侧下肢腱反射(+),右侧下肢腱反射(+),双侧巴氏征阳性。深浅感觉系统查体不配合。颈强直,布氏征、克氏征阴性。既往否认“高血压病、2型糖尿病”病史。辅助检查:葡萄糖10.34 mmol/L,C-反应蛋白99.3 mg/L,纤维蛋白原FBG 5.63 g/L,D二聚体D-Dimer 2300 μg/L ,中性粒细胞比率NEUTA% 89.3%,肝功能:白蛋白ALB 30.8 g/L;粪便常规、大便潜血、甲状腺功能、甲状腺抗体、血脂7项、同型半胱氨酸血症、尿常规、肾功能、电解质、心肌酶谱、叶酸、维生素B12、BNP、脂蛋白磷脂酶A2、血氨、血乳酸均未见明显异常。类风湿因子、抗O:未见明显异常。抗心磷脂抗体:阴性。术前8项:均阴性。肿瘤指标:NSE升高19.89 ng/ml;甲胎蛋白、CEA、CA199、CA125、CA153、CA724、CY211、SCC:未见明显异常。风湿免疫指标:抗核抗体、抗蛋白酶3抗体、抗ENA抗体、免疫球蛋白3项、抗角蛋白抗体、RA33、抗环瓜氨酸肽:均阴性。心电图:未见异常。肌电图提示:双尺神经、双胫神经运动支轻度脱髓鞘;右正中神经CMAP 波幅下降;左尺神经F波未引出,右尺神经、双正中神经F 波出现率下降,双胫神经F 波潜伏期延长;双胫神经H 反射潜伏期延长。腰椎穿刺术:颅内压240 mmH2O;常规:白细胞0.013×106/L;脑脊液蛋白定量159.22 mg/dl。血清、脑脊液副肿瘤抗Ri抗体IgG均为阳性,余脑脊液、血液自身免疫性脑炎、寡克隆带、神经节苷脂抗体谱(抗GQ1B抗体)均为阴性。胸部CT提示(见图1):(1)右上肺病变-考虑恶性肿瘤性病变;(2)右上肺后壁胸膜局部结节影-考虑转移可能性大。患者转入我科后行头部MRI+MRA+CEMRA+增强MRI均未见明显异常(见图2)。结合患者症状、体征、腰穿检查及肌电图检查,考虑吉兰-巴雷综合征可能,转入当天立即给予丙种球蛋白应用5 d,联合营养神经、改善循环、脱水降颅压等对症治疗。患者上述症状未见明显好转或加重。入院第21天(2019年7月16日)早上7:10时患者突然出现癫痫大发作,意识丧失,双眼上翻,牙关紧闭,四肢强直收缩,给予静推安定后逐渐缓解,持续约5 min。当天夜里19:54时患者出现意识不清,昏睡,血氧饱和度下降至82%,心率为114 次/min,呼吸急促,吸痰后血氧饱和度持续不升,患者为呼吸衰竭,危及生命,与患者家属沟通后拒绝转入神经重症监护室,要求出院。
2 讨 论
患者中年男性,急性起病,定位考虑多组颅神经损伤:双侧面神经、舌咽神经、迷走神经、脑干、周围神经损害。定性诊断:患者双侧面神经、四肢肌力下降、腱反射减退,考虑周围神经根病变,脑脊液示蛋白-细胞分离,肌电图示四肢多发周围神经运动支脱髓鞘疾病,考虑吉兰-巴雷综合征(GBS)可能性较大。结合患者既往长期肺癌病史,血清和脑脊液副肿瘤抗Ri抗体IgG阳性,发病过程中迅速出现神志嗜睡-昏迷,脑干颅神经、脑桥侧视中枢、双侧皮质脊髓束损伤,综合考虑诊断为副肿瘤性脑干脑炎合并吉兰-巴雷综合征。
GBS是一种自身免疫介导的周围神经病,主要损害多数脊神经根、周围神经及脑神经的疾病。GBS的病程从轻度到重度残疾,伴有四肢瘫痪、呼吸衰竭及植物自主神经功能紊乱,在发达国家死亡率为2%~3%[1]。用免疫球蛋白(IVIg)或血浆交换(PE)治疗可加速恢复[2]。GBS 的诊断主要依赖于其典型的临床特征:病程多为单项病程,急性起病,病前1~3 w常有呼吸道、胃肠道感染或疫苗接种史,四肢对称性弛缓性瘫痪,肢体末端出现手套-袜样感觉障碍,腱反射减弱或消失,脑脊液呈蛋白-细胞分离和神经传导电生理的的典型改变[3]。结合该患者症状及辅助检查,考虑诊断GBS明确。GBS最早是1916年由Guillain和Barre首先报道,因此命名为吉兰-巴雷综合征。2014年国际最新更新的GBS的诊断标准中,详尽的指出临床分型,主要分为GBS和Miller-Fisher综合征(MFS)两大类,其中MFS分为两大类即:不完全性的MFS和Bickerstaff脑干脑炎(Bickerstaff's brainstem encephalitis,BBE)。不完全性MFS主要临床表现为眼肌麻痹、共济失调、反射减弱或消失,无肢体无力和嗜睡症状;Bickerstaff脑干脑炎主要的临床表现为嗜睡、眼肌麻痹及共济失调,而无肢体乏力,如无眼肌麻痹,则为不完全性的BBE,称为急性共济失调嗜睡型[3]。2003年Odaka等[4]指出MFS、BBE综合征具有相同的自身抗体-抗GQ1b-lgG抗体,研究指出GBS、MFS、BBE三者同属于一个疾病谱,而MFS、BBE是GBS的两种特殊的变异类型,表明BBE和GBS可以重叠,提出MFS、BBE、GBS是一种病因相同、临床表现不同的连续性疾病谱,将其统称为抗GQ1b-lgG抗体综合征[5,6]。最新指南指出,MFS和BBE的诊断无需肢体无力,但需具有眼外肌麻痹和共济失调,两者的区别点是MFS有腱反射减弱/消失而无嗜睡,而BBE有嗜睡而无腱反射减弱/丧失,并且大部分患者MRI检查可见到明显的脑干T1低信号,T2高信号的影像学表现。头部MRI显示脑干异常表现及抗GQ1bIgG抗体阳性为诊断BBE的有力证据[7]。本研究患者虽然出现脑干损伤相关症状,但患者头部MRI未见到明显的脑干病变,血液及脑脊液神经节苷脂抗体谱(抗GQ1b抗体)均为阴性,与BBE诊断不符合,可排除BBE的诊断。
此患者出现典型的中枢神经系统受累的症状,定位于脑干损伤明确。研究指出,脑干脑炎多急性或亚急性起病,其病变局限于脑干或以脑干为主,也可累及邻近组织器官,临床表现多为双侧起病,以多颅神经损害、小脑征及长椎体束征为突出表现。脑干脑炎的病因可分为3种,分别为感染性、自身免疫性和副肿瘤性综合征[8]。引起脑干脑炎的神经系统副肿瘤综合征(paraneoplastic neurological syndrome,PNS)与抗-Yo、抗-Th、抗-Hu、抗-Ri、抗-Ma和抗-amphiphysin抗体有关。结合本研究患者既往长期肺癌病史,血清及脑脊液副肿瘤抗Ri抗体IgG均为阳性,考虑患者此次为副肿瘤相关的脑干脑炎。
PNS是癌肿对神经系统的远隔效应,而非癌肿直接侵犯及转移至神经、肌肉或神经肌肉接头的一组综合征。它既不包括肿瘤对组织的直接压迫、浸润,也不包括手术、应用免疫抑制剂、放疗或化疗的副作用及肿瘤或治疗中引起的机会性感染造成的神经系统损伤[9]。PNS引起的临床症状复杂,既可出现周围神经肌肉的改变,又可出现中枢神经系统各个部位损伤的症状,在临床中大约有20.6%的患者可先出现原发灶症状以后出现中枢神经系统症状,其病程及严重程度与原发性恶性肿瘤的病程和恶性程度无关。
PNS是肿瘤神经交叉反应后的结果,由于肿瘤细胞和神经细胞都可能表达相同的抗原,因此癌症可刺激某些与神经组织抗原交叉反应的抗体产生。与这些抗体相关的主要恶性肿瘤包括妇科癌和乳腺癌,在副肿瘤性小脑变性中可以检测到抗-Yo和抗-Tr抗体[10];乳腺癌和妇科癌与小细胞肺癌抗Ri相关;小细胞肺癌可产生多种PNS的临床综合征,其发病率较高,约3%~5%的小细胞肺癌患者会出现中枢神经系统的损害,包括脑干脑炎、斜视性阵挛-肌阵挛、感觉运动神经病等[11]。PNS的临床分型种类较多:包括小脑变性、脑炎、视网膜病变、脊髓病变、运动、感觉、自主神经、周围神经、神经肌肉接头(Lambert-Eaton肌无力综合征、重症肌无力)、肌肉疾病(多发性肌炎、皮肌炎)等。其中,脑干脑炎并不是神经系统副肿瘤综合征的典型的临床表现[12]。其主要表现为眩晕、吞咽困难、眼球震颤、核间性或核上性眼肌麻痹、斜视眼阵挛、听力减退、共济失调等。并且患者的脑脊液及血清中可出现抗Hu抗体、抗Ma2抗体或抗Ri抗体阳性[13]。其中,抗Ri抗体阳性的PNS脑干脑炎患者,头部MRI通常是正常的,只有少数患者晚期MRI可能出现异常改变,包括小脑蚓部萎缩[14]、中脑背侧异常信号、双侧对称性的脑桥背侧异常信号等。PSN患者的CSF 检查可能显示蛋白质正常或稍升高,但细胞计数通常会增加,淋巴细胞稍高约67%[15]。PNS发病率较低,仅发生于约1%的肿瘤患者,属于罕见病,临床表型复杂多样,且神经系统症状常早于肿瘤的发现,此类患者易漏诊或误诊,因此诊断非常困难。根据2008年Dalmau J 等[16]发表在Lancet Neurol杂志上的诊断标准分为两大类:(1)明确的PNS满足以下4项中的一项:①典型的临床综合征和5 y内发现的相关肿瘤;②有非典型临床综合征和肿瘤的证据,非免疫抑制手段治疗肿瘤后此临床综合征明显改善;③有非典型临床综合征和5 y内发现肿瘤证据以及相关抗体阳性。④有典型或非典型临床综合征及阳性的以及明确的经典抗神经元抗体(Hu,Yo,Ri,CV2/CRMPS,Ma2,抗-amphiphysin)。(2)可能的PNS满足以下3项任一项:①典型临床综合征及高危肿瘤风险(未明确肿瘤且抗体阴性);②典型或非典型临床综合征及目前明确的抗体阳性;③非典型临床综合征及2 y内发现的肿瘤但无抗神经抗体。根据此诊断标准,本研究患者诊断PNS脑干脑炎明确。目前诊断PNS缺乏有效的治疗手段,对患者早识别、早发现,针对原发癌肿进行肿瘤切除、放疗和化疗,或者应用免疫治疗,包括应用糖皮质激素、免疫抑制剂及血浆置换等,能够改善部分患者的神经系统症状。
综上所述,在我们临床诊治过程中,如遇到中老年患者,慢性或亚急性起病,出现累及一个或多个部位的神经系统损害,找不到明确的病因,并且病情逐渐进展,影像学未见明显新发病灶的患者,需警惕肿瘤,须考虑到中枢神经系统副肿瘤综合征的诊断。目前,临床上关于GBS合并PNS脑干脑炎的疾病非常少见,临床医师需认识并了解到此病。通过对临床上更多的新的诊断病例的总结,期待更多的有关GBS合并PNS的疾病分类、发病机制、诊断及治疗方面的进一步研究。

图1 胸部CT(A~C)示:右上肺恶性肿瘤性病变,右上肺后壁胸膜局部结节影
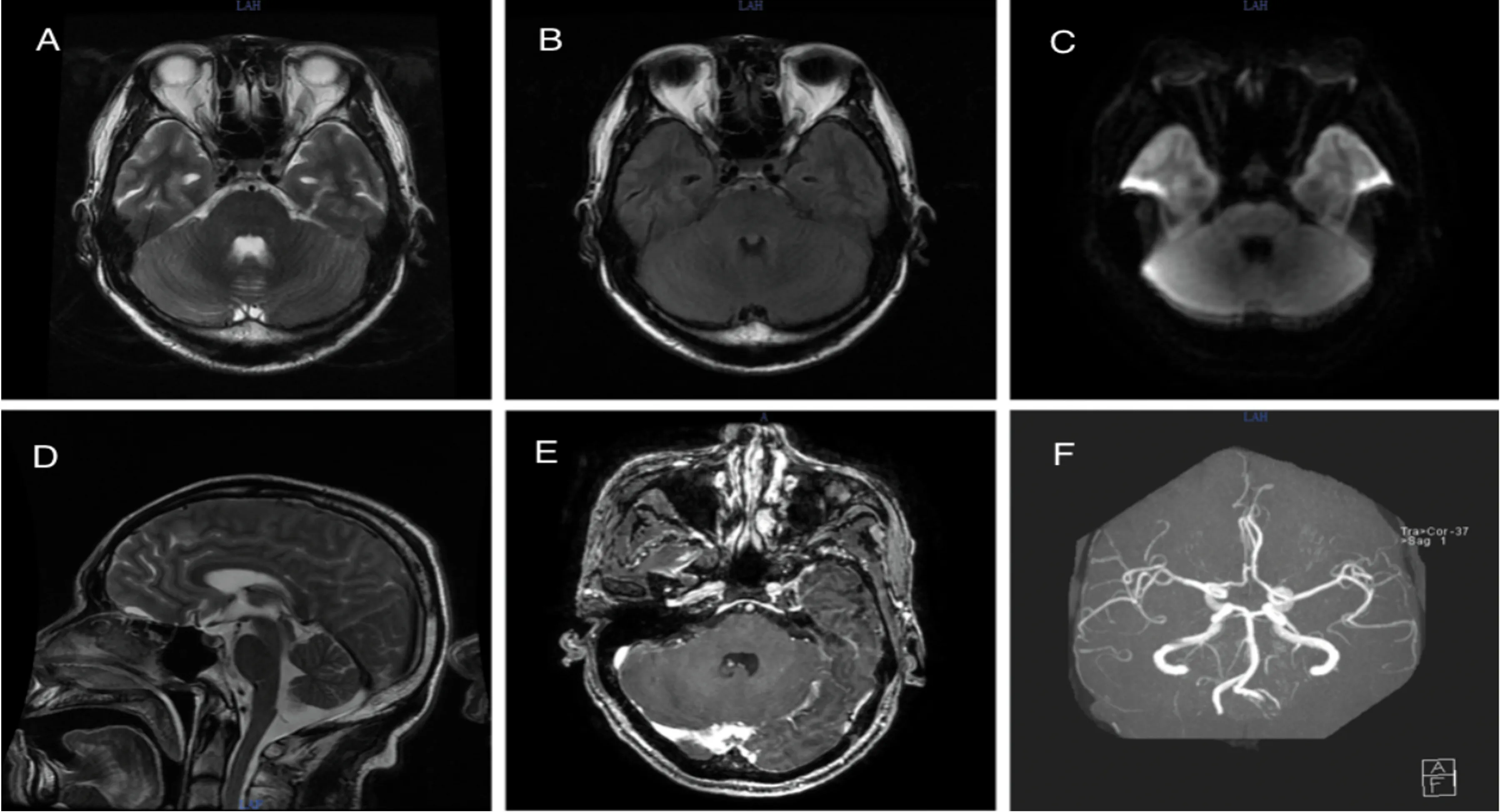
图2 头部MRI平扫+增强+MRA(A~F):脑干未见明显异常;血管未见明显异常
猜你喜欢
现代临床医学(2022年1期)2022-02-12
昆明医科大学学报(2021年12期)2021-12-30
中国听力语言康复科学杂志(2021年6期)2021-12-21
中国听力语言康复科学杂志(2021年6期)2021-12-21
云南医药(2021年3期)2021-07-21
中国现代神经疾病杂志(2021年6期)2021-01-02
云南医药(2020年5期)2020-10-27
科教新报(2019年26期)2019-09-10
家庭科学·新健康(2019年1期)2019-03-06
