
Gitelman综合征1例并文献复习
2021-05-23 13:44:44刘良姝申虎威
山西医科大学学报 2021年4期
刘良姝,耿 艳,邢 莉,申虎威
(长治医学院附属和平医院内分泌科,长治 046000;*通讯作者,E-mail:gengyan2004@163.com)
Gitelman综合征(Gitelman syndrome,GS)又称家族性低钾低镁综合征,以低血钾、低血镁、低尿钙、代谢性碱中毒、肾素-血管紧张素-醛固酮系统(RAAS)激活及正常或偏低血压为主要表现,是一种常染色体隐性遗传的失盐性肾小管疾病,由美国医生Gitelman于1966年首次报道[1,2];通常于青少年或成年时发病,全球患病率约为(1-10)/40 000,在亚洲可能更高[3]。GS的临床表现缺乏特异性,且起病较隐匿,易漏诊和误诊,对临床疑似GS的患者应进行基因检测以明确诊断。现就长治医学院附属和平医院内分泌科2020年8月收治的1例GS患者进行报道,同时对该病进行文献复习。
1 病例报告
1.1 病例资料
患者,女性,15岁,主因“四肢肌肉疼痛半年,反复四肢无力4月”于2020年8月31日入院。该患者半年来常感四肢肌肉疼痛,近4月反复出现四肢无力,于外院就诊查血钾小于2 mmol/L,给予口服氯化钾溶液后症状可改善,自行停止补钾后症状再现,为进一步诊治来我院,门诊查血钾2.18 mmol/L,以“低钾血症查因”收入内分泌科病房。患者自发病以来,时有双手抽搐及四肢麻木,夜尿1-3次/d。否认特殊药物长期服用史,其父母非近亲结婚,无类似疾病家族史。
入院体格检查:体温36.4 ℃,脉搏91次/min,呼吸20次/min,血压110/74 mmHg(1 mmHg=0.133 kPa),身高150 cm,体质量32 kg,体质量指数14.2 kg/m2;神清,反应稍迟钝,甲状腺无肿大,双肺呼吸音清,未闻及干湿啰音,心率91次/min,律齐。腹软,无压痛及反跳痛,双下肢无水肿,四肢肌力5-级,肌张力正常,双侧病理征阴性。
入院辅助检查:血、尿电解质检查结果见表1。血气分析:酸碱度7.54(参考值范围7.35-7.45),二氧化碳分压28.0 mmHg(参考值范围35.0-45.0 mmHg),实际碳酸氢根23.9 mmol/L(参考值范围22.0-26.0 mmol/L),标准碳酸氢根26.7 mmol/L(参考值范围22.0-26.0 mmol/L),实际碱储2.6 mmol/L(参考值范围-2.0-2.0 mmol/L);甲状腺功能七项:促甲状腺激素5.97 μIU/ml(参考值范围0.3-5.0 μIU/ml),其余均正常;促肾上腺皮质激素及皮质醇节律正常;醛固酮174.80 pg/ml(参考值范围70-300 pg/ml),血管紧张素Ⅱ358.70 pg/ml(参考值范围50-120 pg/ml),肾素活性:25.90 ng/(ml·h)[参考值范围0.10-6.56 ng/(ml·h)];氯离子排泄分数:3.37%;氢氯噻嗪试验:氯排泄分数最大差值0.67%;心电图、甲状腺及肾血管彩超未见明显异常。
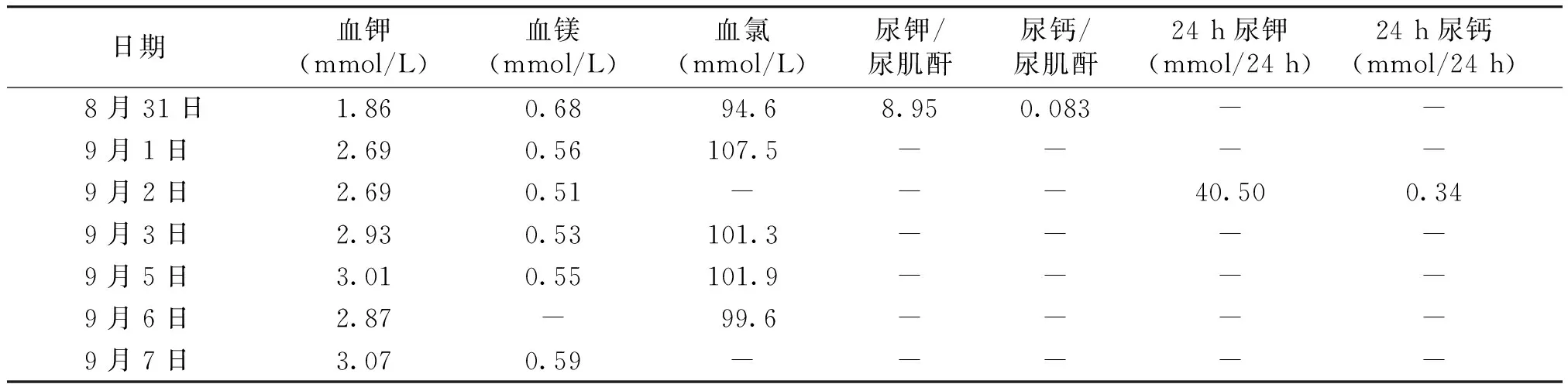
表1 患者住院期间血尿电解质检测结果
1.2 基因检测
经患者及其父母同意后采集外周血送至杭州迪安医学检验中心,提取外周血基因组DNA后用第二代测序技术对患者进行致病基因筛查,共检测20 000个遗传性疾病基因组合外显子及其邻近±10 bp内含子,再用Sanger测序法进行突变位点及突变来源验证。结果显示患者携带SLC12A3基因突变,存在2个变异,分别为c.488C>T(p.Thr163Met)和c.2747+2T>G(见图1,2)。SLC12A3-ex3 c.488C>T(p.T163M),杂合错义突变,该变异已被ClinVar数据库收录,用SIFT和Polyphen-2软件对其蛋白功能进行预测,结果均为有害,因此判定为致病突变;经Sanger验证,该变异来源于母亲。SLC12A3-ex23 c.2747+2T>G,杂合剪接突变,ClinVar数据库尚未收录该变异,经Sanger验证,该变异来源于父亲(见图1,2)。
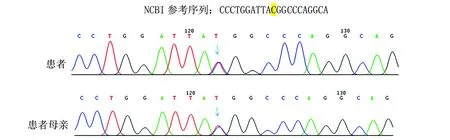
图1 患者及其母亲Sanger测序验证检出SLC12A3-ex3 c.488C>T(p.T163M)
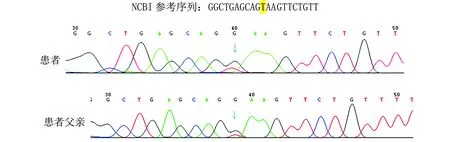
图2 患者及其父亲Sanger测序验证检出SLC12A3-ex23 c.2747+2T>G
1.3 治疗及预后
住院期间药物治疗情况见表2。出院随访4个月,患者未再出现四肢无力,定期复查血钾维持在2.8-3.0 mmol/L,螺内酯片加量至60 mg。

表2 患者住院期间给药情况
2 讨论
GS是一种常染色体隐性遗传性肾小管疾病,是由位于染色体16q13的SLC12A3基因突变所致,该基因可编码肾远曲小管的噻嗪类敏感的钠氯协同转运蛋白(Na-Cl cotransporter, NCCT)。SLC12A3基因突变导致NCCT的结构和/或功能异常,从而引起肾远曲小管对钠离子和氯离子重吸收功能下降,使钾离子和氢离子分泌增加、血容量减少而导致RAAS激活、低血钾和代谢性碱中毒[4]。GS引起低尿钙和低血镁的机制尚不明确,可能是由于氯离子大量外流导致肾远曲小管上皮细胞极性增强,钙离子重吸收增加,尿钙减少;钠离子重吸收障碍导致镁离子重吸收减少,血镁降低[5]。另有研究认为,低血镁的发生可能与瞬时受体电位通道M6(TRPM6)表达下调有关,低尿钙的发生可能与低血容量导致肾近曲小管对钙离子的重吸收增加有关[6]。
GS最常见的症状包括肌肉无力、夜尿增多、感觉异常、麻木等[3],由于其症状缺乏特异性,临床诊断更多依赖于实验室检查,而最终确诊标准为SLC12A3基因中发现2个致病突变[3,7]。本例患者有相关临床症状,存在低血镁及顽固性低血钾,随机尿中尿钾/尿肌酐及24 h尿钾与同步血钾均提示肾性失钾,随机尿中尿钙/尿肌酐及24 h尿钙均提示低尿钙,血浆肾素活性高,氯离子排泄分数大于0.5%,监测血压正常,肾脏超声未见明显异常,否认利尿剂及缓泻剂应用史,均支持GS诊断,尚需与另一种常染色体隐性遗传性肾小管疾病Bartter综合征(Bartter syndrome, BS)相鉴别。BS发病更早,血镁和尿钙不低,氯离子清除试验(氢氯噻嗪/呋塞米试验)有助于鉴别诊断,基因检测可明确诊断。本例患者行氢氯噻嗪试验示氯排泄分数最大差值小于2.86%[8],故临床诊断为GS。为明确诊断,我们采集患者及其父母的外周血进一步行基因检测,结果示检出1个致病变异和1个疑似致病变异,分别来源于患者母亲及父亲。
目前已报道的GS相关基因SLC12A3突变位点约488个,包括错义突变、剪接突变、无义突变、移码突变等,其中大多数为错义突变,且复合杂合突变比纯合突变更常见,大部分患者的两个等位基因上有两个不同的突变[9]。错义突变T60M是中国患者中最常见的突变[10],而剪接突变IVS9+1G>T是欧洲患者中最常见的突变[11]。本例患者SLC12A3基因有两个突变位点,一个为错义突变T163M,来源于母亲;另一个为剪接突变c.2747+2T>G,来源于父亲,属于复合杂合突变。错义突变T163M可将NCCT第163位氨基酸的苏氨酸变为蛋氨酸,已报道的该位点既有复合杂合突变也有纯合突变,且前者多于后者[4,10]。对于剪接突变c.2747+2T>G,目前仅有一项来自中国的队列研究报道了该剪接突变位于高度保守的剪接信号AG或GT中,用NNSplice,SpliceView和NetGene2软件进行生物信息学分析,结果显示该变异可能影响剪接并具有致病性,推测它可能会使终止密码子提前出现或读码框移码,并导致mRNA加工不当或不稳定,从而造成NCCT功能失活或缺陷[12]。根据常染色体隐性遗传规律、GS的确诊标准及生物信息学分析,我们推测该剪接突变为致病变异的可能性极大,尚需进行功能验证以明确。
GS的临床表型异质性较大,有的患者可能没有任何症状,有的患者则有低钾血症的相关症状,携带相同突变的患者可能表现不同的症状,纯合子患者的症状比杂合子患者更严重,男性患者的症状比女性患者更严重[4]。另外,血镁水平可能提示病情的严重程度,血镁水平低的患者症状往往更严重[5,13]。目前研究认为GS的基因型和表型之间无明显相关性,尚需进一步研究以明确。
GS的治疗应个体化,其主要治疗方法是终身口服补钾和/或镁,血钾的目标值为3.0 mmol/L,血镁的目标值为0.6 mmol/L;存在低镁血症时,应该首先考虑补充镁,因为补镁有利于血钾水平的提高,并可降低手足抽搐和其他并发症的风险[3]。多数症状在补钾和/或镁后得到改善,对于持续有低血钾症状的患者,建议使用保钾利尿剂,如阿米洛利、螺内酯、依普利酮等[3,14,15]。依普利酮属于选择性的醛固酮受体拮抗剂,其副作用少于螺内酯,但有研究报道螺内酯的补钾效果可能优于依普利酮,且口服补钾联合螺内酯可更有效地提高血钾水平[16]。本例患者于外院经过口服补钾后症状改善,自行停止补钾后症状加重,于我院住院后予以静脉及口服补钾、口服补镁及螺内酯片治疗后,出院前复查血钾及血镁接近目标值,但出院后在螺内酯片加量的情况下,复查血钾水平较前稍下降,建议患者适当增加门冬氨酸钾镁片的剂量,但需注意观察有无腹泻、呕吐等不良反应。GS总体预后良好,对于确诊患者,需进行详细的家系调查[7],但患者爷爷奶奶、外公外婆及其同胞弟弟因均未出现相关临床症状及家庭经济较困难,拒绝行基因检测。本例患者存在低钾血症的相关症状,实验室检查支持GS诊断,基因检测明确GS诊断,联合口服补钾补镁及螺内酯片治疗后效果尚可。该患者处于青春期,生长迟缓,在随访过程中除了对病情进展及可能的并发症进行评估外,还应进行生长发育状况的评估。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
中外医疗(2022年31期)2022-04-03 06:42:44
透析与人工器官(2020年1期)2020-11-16 01:42:28
养生大世界(2019年12期)2019-12-11 10:08:01
家庭科学·新健康(2018年8期)2018-10-30 10:23:20
中国社区医师(2017年34期)2018-01-06 00:53:12
护理实践与研究(2015年1期)2015-08-05 03:36:50
保健与生活(2014年3期)2014-04-29 00:44:03
