
ARID1B 基因变异致Coffin-Siris 综合征1 例报告并文献复习
2021-05-01 11:21李红英
临床儿科杂志 2021年4期
徐 欣 汤 健 张 丽 陆 芬 李红英
南京医科大学附属儿童医院康复科(江苏南京 210008)
Coffin-Siris综合征(Coffin-Siris syndrome,CSS,OMIM 135900)是一种罕见的常染色体显性遗传疾病,主要表现为智力运动发育迟缓、多毛症、粗糙的面部特征、肌张力低下、指骨/指甲发育不全等[1]。因Coffin 和Siris 于1970 年首次报道而命名。目前国际上已明确的CSS 致病基因有9 种,分别为编码BAF(Brg-1 associated factors)染色质重塑复合物的基因ARID 1A、ARID 1B、ARID 2、SMARCA4、SMARCB1、SMARCE1、DPF2以及参与BAF复合物转录调控的基因SOX11、PHF6[2-5]。CSS尚无明确的发病率报道。到目前为止,国外报道200余例,中国人群中只有散在数例报道[6-9]。本文报告1例ARID1B基因变异致CSS患儿,并结合相关文献,总结其临床表型和基因变异特征。
1 临床资料
患儿,男,2岁8月龄,因智力、运动发育落后1年余入院。患儿系G3P1(G1、G2系人工流产),足月剖宫产(羊水浑浊),出生体质量3 kg,Apgar评分不详,无窒息缺氧史。患儿生后吃奶差,身长、体质量增长慢;大运动发育落后于同龄儿童,6个月会翻身,12个月独坐,18个月独走。1岁10个月因“右侧隐睾、右侧腹股沟疝”行手术治疗。入院时患儿独走步态蹒跚,步基大,步幅小,膝过伸,双足扁平、外翻,下蹲后不能起立,不能跑、跳,不能上楼梯;会叫爸妈,只会说3、4字的简单词语,不能完成简单指令,眼神交流少,喜独自玩耍。患儿父母非近亲结婚,家族中无类似疾病者。入院体格检查:体质量12.5 kg(P10~P25),身高93 cm(P25~P50),头围49 cm;毛发稀疏,特殊面容,前额发际线低,拱形眉,眉毛浓密,长睫毛,鼻翼宽,鼻梁低,上嘴唇薄,下嘴唇厚且外翻,唇毛明显(图1A);心肺腹无异常;四肢肌张力低,膝腱反射无异常,病理反射未引出;脊柱无畸形,右足趾甲小(图1B)。实验室检查:血、尿、粪常规,肝肾功能,电解质,心肌酶谱,血脂未见异常;甲状腺功能无异常;血串联质谱未见异常。头颅磁共振成像(MRI)未见异常;双手正位片、骨盆正位片无异常;脑电图、视觉听觉诱发电位未见异常;心电图示窦性心动过缓伴不齐;心脏B 超无异常。0~6 岁儿童Gesell发育诊断量表测试示大运动相当于15个月,适应性、精细动作、语言、个人-社交相当于18个月。
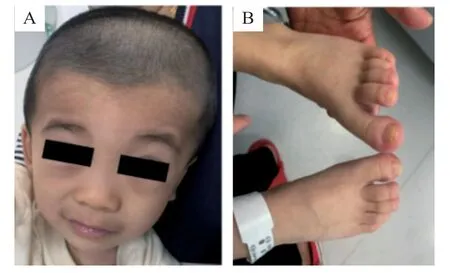
图1 患儿面容以及双足外观
为进一步明确病因,经医院医学伦理审核以及家长知情同意,抽取患儿及父母静脉血2 mL,送广州金域(医学检验)公司进行全外显子测序。对基因组DNA 文库进行人类全外显子捕获,在MiSeq 测序仪(Illumina Inc,USA)测序平台进行高通量测序,测序数据经生物信息学分析及数据库注释,并按照美国医学遗传学与基因组学会(ACMG)指南[10]对变异位点进行致病性分析。结果发现,患儿ARID 1 B基因(NM_020732.3)第20 外显子存在1 个杂合错义变异c.6257 T>C(p.Leu 2086 Pro),第6257 位碱基由胸腺嘧啶变异为胞嘧啶,导致该基因编码的第2086 氨基酸由异亮氨酸(Leu)变成脯氨酸(Pro)。对患儿及其父母目标序列进行Sanger 测序验证。扩增ARID 1 B 基因第20 号外显子的引物序列,正向引物5'-AAAGCATCATAGCAACCATCG-3’;反向引物5'-TGGGCAAGGTTCGATAAAAG-3’。验证结果示患儿ARID1B基因存在杂合变异,父母均不携带该变异,属于新发变异(图2)。按照ACMG指南对该变异进行分析:①为新生突变(PS 2);②比照千人基因组数据库(1000 Genomes)、人类基因变异数据库(HGMD)未见变异报道和收录(PM2);③检索相关文献,在CSS患儿中检测到ARID1B基因c.6257T>G(p.Leu2086Arg)变异[11](PM5);④生物信息学软件(Polyphen2、SIFT、Mutation Taster)预测均为有害。应用HomoloGene系统对人类、猕猴、狼、牛、大鼠、小鼠、鸡、斑马鱼、假丝酵母的ARID1B蛋白保守性分析发现,ARID1B第2086位Leu在无脊椎动物、脊椎动物以及哺乳动物之间均高度保守(PP3)(图3) 。根据ACMG指南评级规则,ARID1B基因c.6257T>C(p.Leu2086Pro)变异评定为“可疑致病”(PS2+PM2+PM5+PP3)。结合患儿临床表现,患儿最终诊断为CSS。
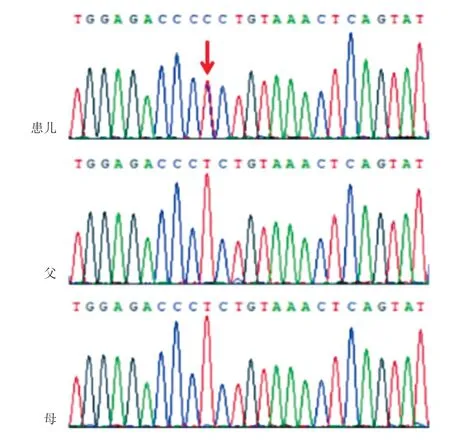
图2 患儿及父母ARID1B 基因Sanger 测序图

图3 ARID1B 蛋白第2086 位氨基酸保守性分析图
2 讨论
CSS绝大多数病例为新生变异所致,多为散发发病,其临床表型多样,特殊的面容与多系统异常表型对诊断具有非常重要的价值。CSS主要临床表现为智力运动发育迟缓、粗糙的面部特征、头皮毛发稀疏、多毛症、肌张力低下、第五指指骨或指甲发育不全。
以“Coffin-Siris综合征”、“Coffin-Siris syndrome”、“ARID 1 B”为关键词,对万方数据库、中国知网及PubMed数据库自建库时至2020年9月收录的文献进行检索,共检索到59 篇文献,其中临床资料齐全、经基因检测确诊的英文文献 27篇、中文文献2篇,排除重复报道的病例,共有CSS 患者86 例[2-3,6,8-18],其中男37 例、女49 例。本例患儿临床特征与已报道病例(表1)基本相符,表现为智力运动发育落后、生后喂养困难、体质量增长不良、典型的面部特征、多毛症、肌张力低下、右足趾甲小、隐睾以及腹股沟疝。CSS最常见的临床特征为智力运动发育落后,程度轻重不等,其中语言发育通常比大运动发育更受影响[13]。其次是多毛症,表现为头皮毛发延伸到前额,面部毛发过度生长。绝大多数患者有颜面部异常表现,较常见的包括低前/后发际线、头皮毛发稀疏、嘴巴宽阔,下嘴唇厚、上嘴唇薄、鼻翼宽、鼻梁低、鼻孔前倾、拱形眉毛,其他少见表现有上眼睑下垂、低耳位、小头畸形、高腭弓、腭裂、悬雍垂裂等。骨骼系统受累常见的异常表现为1个或多个指甲/趾甲发育不全,第五指末节指骨发育不全常见,其他骨骼异常可见关节松弛、脊柱侧弯、漏斗胸、小指内弯等。多数CSS患者生后有喂养困难,严重者需鼻饲管喂养[12]。其他胃肠道问题可表现为便秘、胃食管反流、腹泻等。部分CSS 患者可出现视力受损,常合并斜视、近视、散光、视神经发育不良、眼球震颤、黄斑发育不良等眼部异常。约1/3 的CSS 患者生后逐渐出现身材矮小(<-2 SD),骨龄通常落后于年龄2~3 年[17]。听力受损亦可发生于小部分患者中,多为感音神经性,少数可为传导性。CSS患者癫痫发作的年龄从出生到青少年期不等,主要表现为强直性肌阵挛,另有一些患者脑电图异常但无临床发作。部分CSS 患者可合并泌尿、生殖以及心脏等病变,如肾积水、隐睾、房间隔缺损、室间隔缺损、疝、漏斗胸等[12-13],少数患者可有内分泌异常表现为甲状腺功能减退、糖尿病、生长激素缺乏等。一些CSS患者在青少年期可出现行为问题,包括孤独症谱系障碍、注意力缺陷多动障碍、冲动等。CSS 患者最常见的头颅MRI异常为胼胝体发育不良,少数可见髓鞘化延迟、巨脑池、侧脑室枕角扩大畸形等。
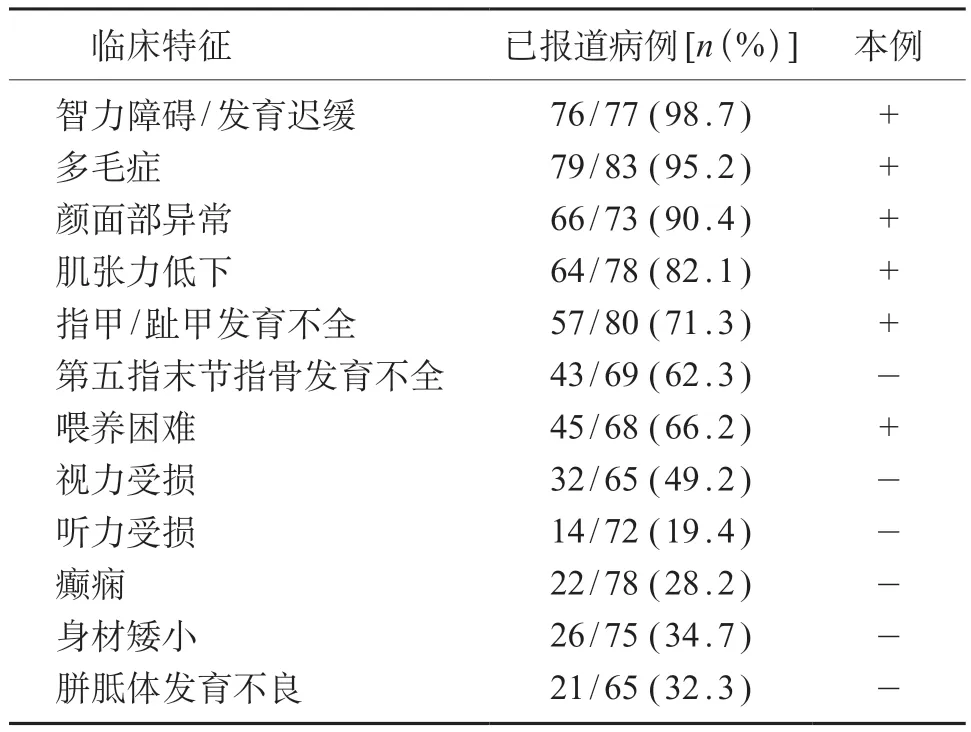
表1 86例ARID1B基因变异致Coffin-Siris综合征患者临床特征
自2012年发现编码BAF复合物亚单位的基因变异可致CSS[3],目前共发现9种与CSS致病相关基因,分别为编码BAF染色质重塑复合物的基因ARID1A、ARID1B、ARID2、SMARCA4、SMARCB1、SMARCE1、DPF2及其下游调控基因SOX11、PHF6。ARID1B是CSS患者中变异频率最高的基因,占68%~83%[3,11,13]。ARID 1 B 基因位于人类第6 号染色体q 25.3 区,包含24个外显子,编码包含2 249个氨基酸的蛋白。AT丰富结合域1 B(AT-rich interactive domain-containing protein 1B,ARID1B)蛋白是BAF染色质重塑复合物的非催化亚基之一,又称BAF250b亚基,包含2个结构域,即富含AT 的相互作用域和C 末端的BAF 250结构域。ARID1B蛋白定位于细胞核,具有与DNA或蛋白质结合能力,可以调控复合物靶向性和ATP酶活性。BAF复合物即哺乳动物SWI/SNF复合物,可通过ATP水解后释放能量来调控染色质动态结构和基因表达,在细胞增殖分化以及神经发育过程中发挥重要作用[19-20]。ARID1B基因表达广泛,人类RNA测序显示其在大脑、皮肤、甲状腺、淋巴结、卵巢、膀胱等部位均有表达,故CSS患者在临床可表现出多系统受累以及临床表型的异质性。研究发现,ARID 1 B 基因参与调节发育中小鼠大脑的树突分化[21]。ARID1B基因杂合变异小鼠可表现出严重的行为异常、缩小的体型以及基因表达的显著变化,上述表现与CSS患者部分表型相似。ARID1B基因杂合变异小鼠由于GABA能神经元数量的改变和抑制性突触传递的受损,表现出明显的皮层兴奋性/抑制性失衡以及异常的表观遗传修饰,包括组蛋白乙酰化和甲基化,从而影响表观遗传调控,尤其是神经发育的调控[22]。
迄今报道的CSS患者ARID1B基因变异类型包括无义变异、插入/缺失变异、剪接位点变异、错义变异以及单外显子、多外显子和全基因缺失[11]。错义变异报道很少,主要集中在BAF250结构域,而无义、缺失/插入、剪切位点变异可分布于各个区域。其中无义变异以及插入/缺失变异约占所有致病性变异的80%,均导致下游形成无义密码子,使翻译提前终止,形成截短型蛋白,影响正常蛋白的功能。所以CSS患者的临床表型是由于ARID1B单倍剂量不足。但在CSS患者中尚不存在特定的基因型-表型相关性。本例患儿全外显子基因检测发现ARID 1 B 基因第20 号外显子存在1个杂合错义变异c.6257T>C,导致氨基酸改变p.Leu2086Pro,该位点位于ARID1B蛋白BAF250结构域中的BC 盒(BC-box),在不同物种间高度保守。体外研究发现,ARID 1 B 蛋白以BC 盒依赖方式与elongin C、cullin 2和Roc1结合形成E3泛素连接酶,以组蛋白H 2 B 为靶点进行单泛素化[23]。BC 盒变异体在体内不稳定,以蛋白酶体依赖方式自泛素化和降解。以上功能研究可能提示本例患儿ARID1B基因变异致病的潜在机制。
CSS 和Nicolaides-Baraither 综合征(Nicolaides-Baraither syndrome,NCBRS)有一部分表型重叠。NCBRS 是由SMARCA 2 基因变异所致,也可表现为粗糙的面部特征、头皮毛发稀疏、智力运动落后,有时难以与CSS 鉴别。但NCBRS 患者的骨骼系统改变多为指节间关节和远端指骨非炎症性突起(高达85%)[24],而少有CSS患者的第五指骨/指甲发育不全。随着SMARCA 2 变异的发现,以前被诊断为CSS的部分病例被重新分类为NCBRS[25]。
目前CSS尚无特效治疗方法,主要是对症和支持治疗。喂养困难或体质量增长不良患儿需经消化科以及营养科评估,可予鼻饲管喂养或补充营养。身材矮小患儿每年需定期监测生长激素以及骨龄,必要时可注射生长激素。合并先天性心脏病或泌尿生殖系统畸形患者需行手术治疗。有癫痫发作的患者需加用抗癫痫药物,定期复查视频脑电图。智力运动落后者可进行包括物理治疗、言语治疗等综合康复治疗。对于有孤独谱系障碍倾向的患儿,可利用相关量表进行评估,达到早期识别和干预的目的。合并眼部病变者建议每年进行眼科检查,包括眼底检查,并根据需要进行视力测试和矫正。听力障碍者可佩带助听器。CSS患儿父母再生育时需进行遗传咨询,因不能排除患儿父母为生殖细胞嵌合携带者可能。
综上,临床上对于有智力运动落后、多毛症、粗糙的面部特征、肌张力低下、指骨/指甲发育不全等患儿应考虑CSS可能,应尽早完善遗传学检查,明确病因,明确诊断。本例CSS患儿ARID1B基因存在新发现的c.6257T>C错义变异,不仅丰富了ARID1B基因变异谱,同时为产前遗传咨询提供了依据。
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
分子诊断与治疗杂志(2022年9期)2022-10-09
中国现代医生(2022年21期)2022-08-22
农村科学实验(2022年2期)2022-03-12
支部建设(2020年15期)2020-07-08
三农资讯半月报(2020年2期)2020-03-09
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
湖北农业科学(2014年11期)2014-09-10
雕塑(1996年4期)1996-07-12
