
板青颗粒质量标准提升研究
2021-04-19 11:27刘建晖石慧慧汪云花
中国兽药杂志 2021年1期
熊 玥,刘建晖,石慧慧,汪云花,孙 瑶
(江苏省兽药饲料质量检验所,南京 210036)
板青颗粒收载于《中国兽药典》2015年版二部,由板蓝根、大青叶两味中药煎煮浓缩后,与适量蔗糖、糊精混合制粒而成[1]。方中板蓝根、大青叶均可清热解毒,板蓝根重在凉血利咽,大青叶偏于凉血消斑,二药合用,共奏清热解毒、凉血之功。临床上广泛用于畜禽风热感冒、咽喉肿痛、热病发斑等温热性疾病的防治[2]。目前板青颗粒的质量标准相对滞后,仅有理化显色鉴别及精氨酸的薄层鉴别,专属性不强,无指标性成分的定性鉴别及定量测定,且精氨酸的薄层鉴别方法在实际检验中存在一定的问题,很难对板青颗粒质量的优劣进行全面评价,导致市场上的板青颗粒质量参差不齐,也给假劣产品混入提供了可乘之机。本研究参考文献报道[3-9]及《中国兽药典》2015年版二部中板蓝根和大青叶药材质量标准项下的有关方法[1],以精氨酸、脯氨酸、亮氨酸、靛玉红为专属性成分,建立薄层色谱鉴别方法;以尿苷、鸟苷、(R,S)-告依春和腺苷为指标性成分,建立高效液相色谱定性及定量测定方法,完善板青颗粒的质量标准,并利用新建立的方法对市售的17批次样品进行了评价性研究,为《中国兽药典》2020年二部中板青颗粒质量标准提升提供了实验数据。
1 仪器与材药
1.1 仪器 半自动点样器(Linomat 5型,瑞士Camag公司);薄层色谱数码成像系统(TLC Visualizer型,瑞士Camag公司);高效液相色谱仪(ACQUITY Arc型,美国Waters公司,配PDA检测器和Empower 3工作站);电子天平(XA105DU型,瑞士Mettler Toledo公司);数控超声波清洗器(KQ-500DV型,昆山市超声仪器有限公司)。
1.2 材料 甲醇(色谱级,德国Merck公司);水为超纯水;其他试剂均为分析纯(南京化学试剂有限公司);硅胶G薄层板(青岛海洋化工有限公司)。精氨酸对照品(批号140685-201707,含量99.9%)、亮氨酸对照品(批号140687-201905,含量99.9%)、脯氨酸(批号140677-201808,含量99.9%)、靛玉红对照品(批号110717-201805,含量99.6%)、尿苷对照品(批号110887-201803,含量99.5%)、鸟苷对照品(批号111977-201501,含量93.6%)、(R,S)-告依春对照品(批号111753-201706,含量100.0%)、腺苷对照品(批号110879-201703,含量99.7%)均购自中国食品药品检定研究院;除自制的3批板青颗粒(批号:20200223、20200304、20200325)外,另收集市售样品17批次(来源批号等信息见表6)。
2 方法及结果
2.1 鉴别
2.1.1 氨基酸的薄层色谱鉴别 取本品研细的粉末1 g,加乙醇10 mL,超声处理30 min,滤过,滤液自然挥发至约2 mL,作为供试品溶液。另取精氨酸对照品、脯氨酸对照品、亮氨酸对照品适量,加乙醇制成每1 mL各含1 mg的溶液,作为混合对照品溶液。照薄层色谱法(《中国兽药典》2015年版二部 附录0502)试验,吸取上述混合对照品溶液2 μL、供试品溶液5 μL,分别点于同一硅胶G薄层板上,以正丁醇-冰醋酸-水(19∶7∶5)为展开剂,展开,取出,晾干,喷以茚三酮试液,在105 ℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,结果见图1。

S.亮氨酸、脯氨酸、精氨酸(从上至下) S1.大青叶阴性对照 S2.板蓝根阴性对照1~5.板青颗粒
2.1.2 靛玉红的薄层色谱鉴别 取本品研细的粉末10 g,加二氯甲烷50 mL,超声处理30 min,滤过,滤液浓缩至约1 mL,作为供试品溶液。另取靛玉红对照品适量,加二氯甲烷制成每1 mL含1 mg的溶液,作为对照品溶液。照薄层色谱法(《中国兽药典》2015年版二部 附录0502)试验,吸取上述对照品溶液5 μL、供试品溶液10 μL,分别点于同一硅胶G薄层板上,以环己烷-二氯甲烷-丙酮(5∶4∶2)为展开剂,展开,取出,晾干。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。结果见图2。
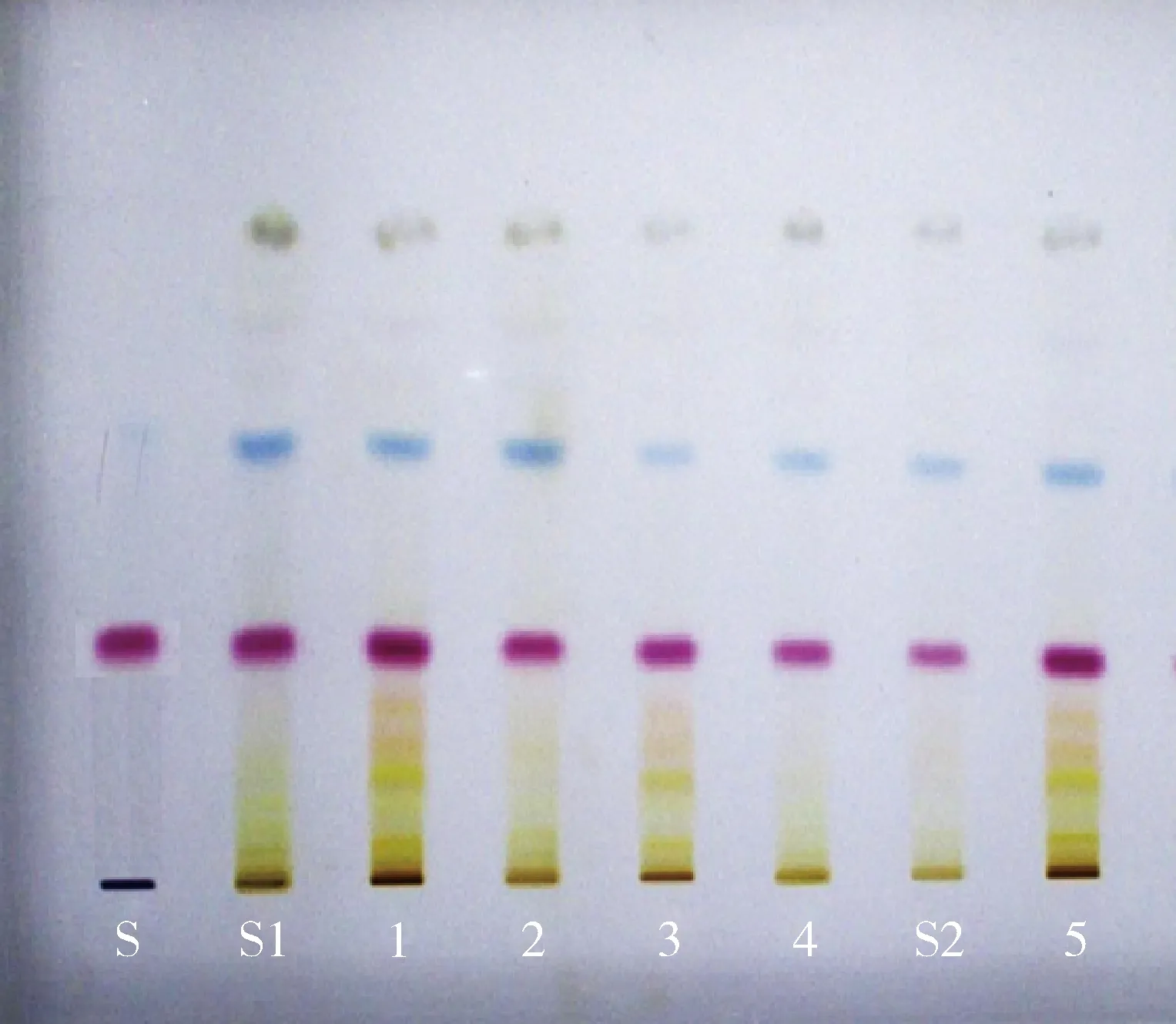
S.靛玉红 S1. 板蓝根阴性对照 S2.大青叶阴性对照1~5.板青颗粒
2.1.3 尿苷、鸟苷、(R,S)-告依春和腺苷的高效液相色谱定性鉴别 取“2.2.3”项下制备的溶液,作为供试品溶液。另取“2.2.2”项下制备的溶液,作为混合对照品溶液。按“2.2.1”项下的色谱条件试验,采用二极管阵列检测器(PDA)进行全波长扫描,记录混合对照品溶液及供试品溶液在210~400 nm的吸收光谱图,提取245 nm波长处的色谱图。结果,在供试品色谱中,呈现与4种对照品色谱峰保留时间一致的色谱峰,且各峰的吸收光谱图与相应对照品一致,见图3。

图3 混合对照品(A)和板青颗粒(B)的高效液相色谱定性鉴别光谱指数图
2.2 4 种指标性成分的定量测定
2.2.1 色谱条件与系统适用性试验 SHISEIDO Capcell Pak C18 MG色谱柱(250 mm × 4.6 mm,5 μm);甲醇(A)-水(B)为流动相,梯度洗脱(0~15 min,3%~5% A;15~20 min,5%~10% A;20~23,10%~13% A;23~38 min,13% A);流速为0.8 min·mL-1;柱温为30 ℃;检测波长为245 nm;进样量为10 μL;理论板数按(R,S)-告依春计算应不低于5000,供试品图谱中尿苷、鸟苷、(R,S)-告依春、腺苷与其他组分均能达到基线分离,见图4。
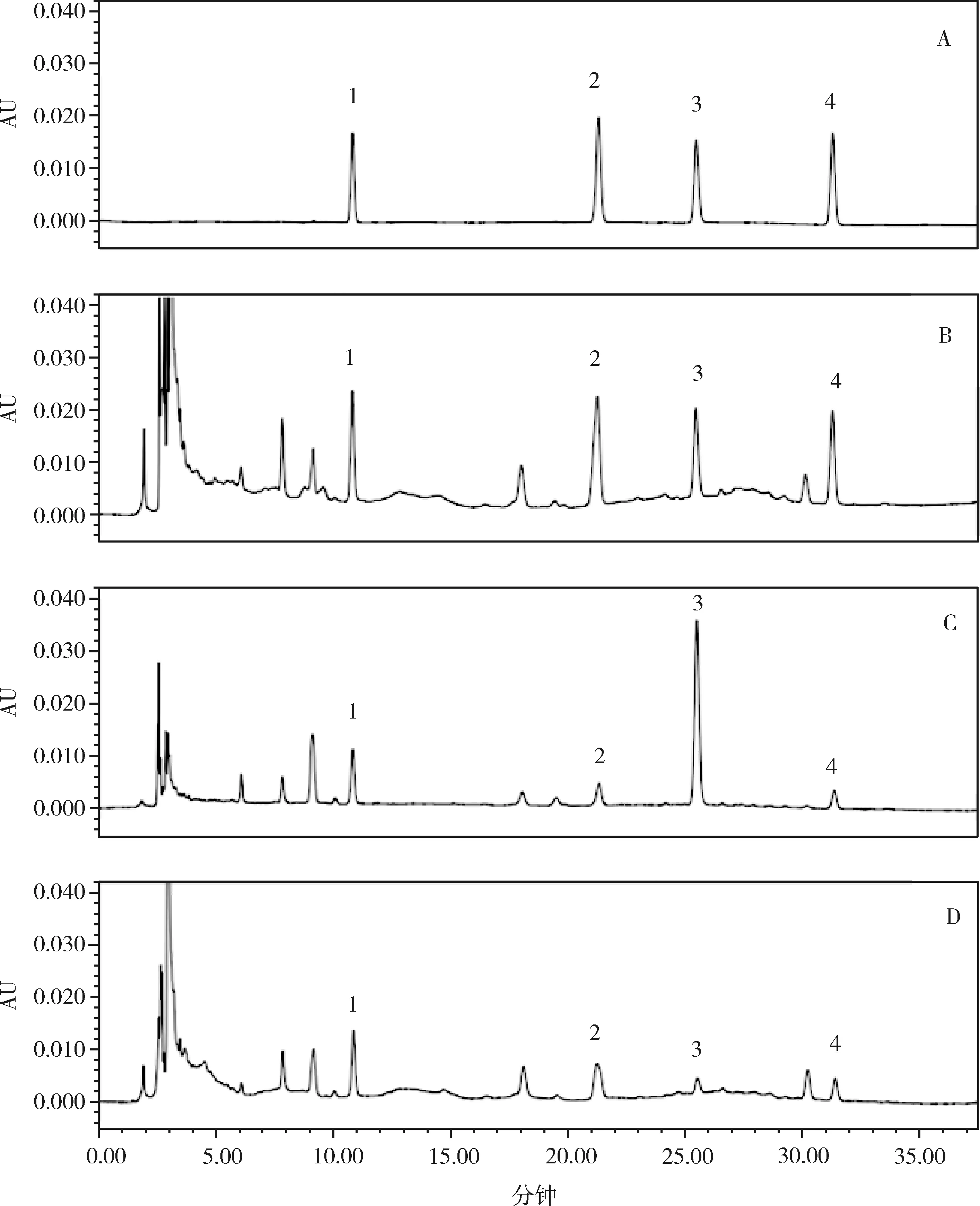
1.尿苷 2.鸟苷 3.(R,S)-告依春 4.腺苷
2.2.2 对照品溶液的制备 取尿苷、鸟苷、(R,S)-告依春和腺苷对照品适量,精密称定,加5%甲醇溶液制成每1 mL含尿苷、鸟苷、(R,S)-告依春和腺苷分别为10 μg、10 μg、3 μg 和10 μg的混合对照品溶液,即得。
2.2.3 供试品溶液的制备 取本品适量,研细,取约2 g,精密称定,置100 mL量瓶中,加5%甲醇溶液70 mL,超声处理(功率500 W,频率40 kHz)20 min,放冷,用5%甲醇溶液稀释至刻度,摇匀,滤过,取续滤液,即得。
2.2.4 线性关系考察 分别取尿苷、鸟苷、(R,S)-告依春和腺苷对照品适量,精密称定,用5%甲醇溶液溶解并稀释制成含尿苷为108.06 μg·mL-1、鸟苷为101.09 μg·mL-1、(R,S)-告依春为31.44 μg·mL-1和腺苷为105.18 μg·mL-1的混合对照品贮备溶液。精密吸取对照品贮备液适量,加5%甲醇溶液分别稀释制成含尿苷32.42、21.61、10.81、5.40、2.16、1.08 μg·mL-1,含鸟苷30.33、20.22、10.11、5.05、2.02、1.01 μg·mL-1,含(R,S)-告依春9.43、6.29、3.14、1.57、0.79、0.39 μg·mL-1,含腺苷31.56、21.04、10.52、5.26、2.10、1.05 μg·mL-1的系列溶液,按拟定色谱条件进行测定,记录峰面积,以对照品的浓度为横坐标(x),以峰面积积分值为纵坐标(y),绘制标准曲线,见表1。
2.2.5 检测限和定量限 精密量取已知质量浓度的对照品溶液,用5%甲醇溶液依次稀释制成浓度由高到低的系列溶液,测得尿苷、鸟苷、(R,S)-告依春和腺苷的检测限(S/N=3)和定量限(S/N=10),用质量单位表示,见表1。
2.2.6 精密度试验 精密吸取混合对照品溶液10 μL,连续进样6次,测得各峰相对保留时间的RSD(n=6)<0.1%,尿苷、鸟苷、(R,S)-告依春和腺苷各峰面积的RSD(n=6)分别为0.4%、0.3%、0.4%和0.4%,方法精密度良好。
2.2.7 重复性试验 取样品(批号20200223)按“2.2.3供试品溶液的制备”项下操作,平行制备6份,按上述色谱条件进样测定,测得每1 g供试品中尿苷、鸟苷、(R,S)-告依春和腺苷的平均含量分别为583、599、150、449 μg,RSD(n=6)分别为0.4%、0.6%、1.0%和0.5%,表明该方法重复性良好。
2.2.8 稳定性试验 取同一份供试品(批号20200223)溶液,分别在0、2、4、8、12、24、48、72 h按上述色谱条件进样,记录供试品溶液中尿苷、鸟苷、(R,S)-告依春和腺苷的色谱峰保留时间及峰面积,各峰保留时间RSD(n=8)<0.1%,峰面积RSD(n=8)分别为0.9%、1.0%、1.6%和0.6%,表明供试品溶液在72 h内稳定性良好。
2.2.9 回收率试验 取已知含量的板青颗粒(批号20200223)细粉约1.0 g,精密称定,置100 mL量瓶中,按供试品已知成分含量的50%、100%、150%三个水平分别加入浓度为尿苷54.03 μg·mL-1、鸟苷50.54 μg·mL-1、(R,S)-告依春15.72 μg·mL-1和腺苷52.59 μg·mL-1的混合对照品溶液5、10、15 mL。按“2.2.3”项下操作并进样测定,计算加样回收率。结果样品中尿苷、鸟苷、(R,S)-告依春和腺苷的平均回收率分别为98.6%、100.5%、98.5%和99.8%,RSD(n=9)分别为1.0%、1.3%、1.1%和1.4%。见表2~5。
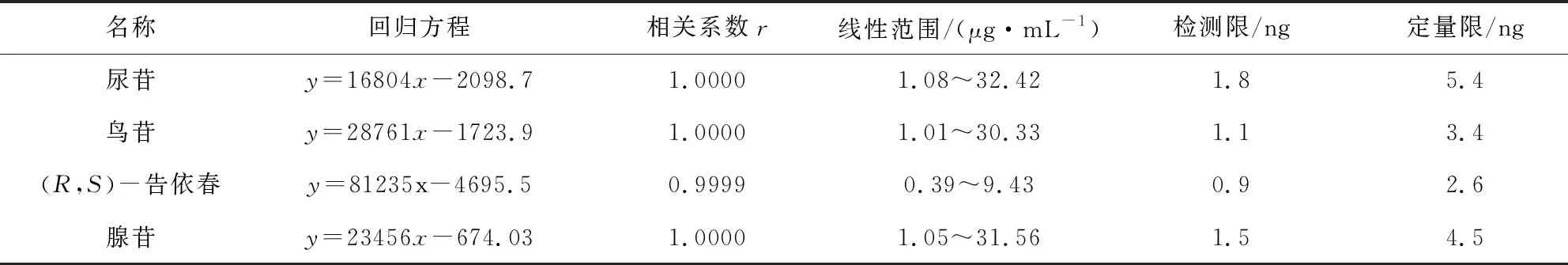
表1 回归方程、线性范围、定量限和检测限
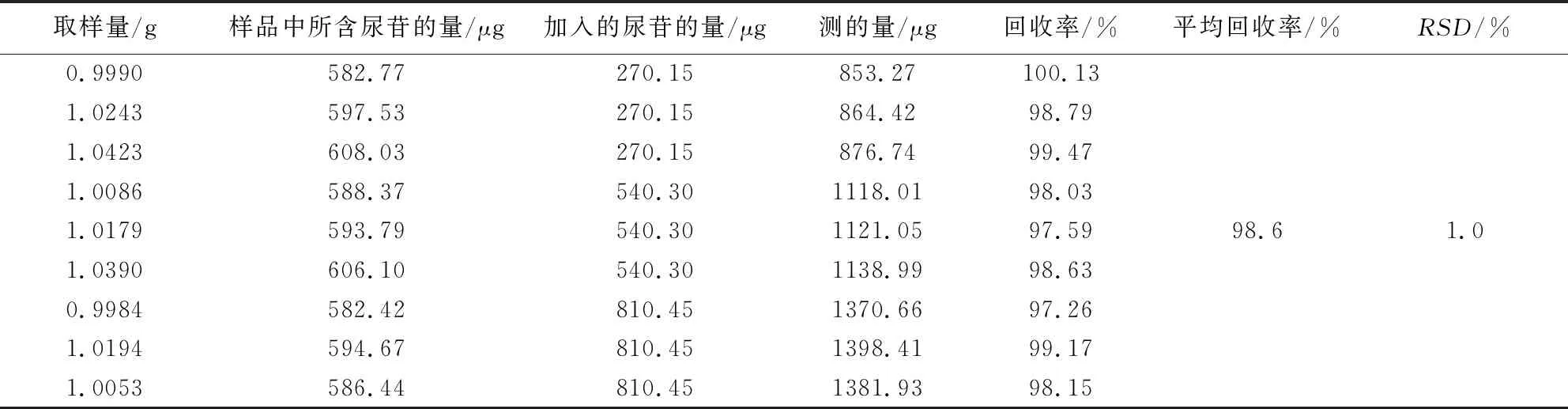
表2 板青颗粒中尿苷加样回收率
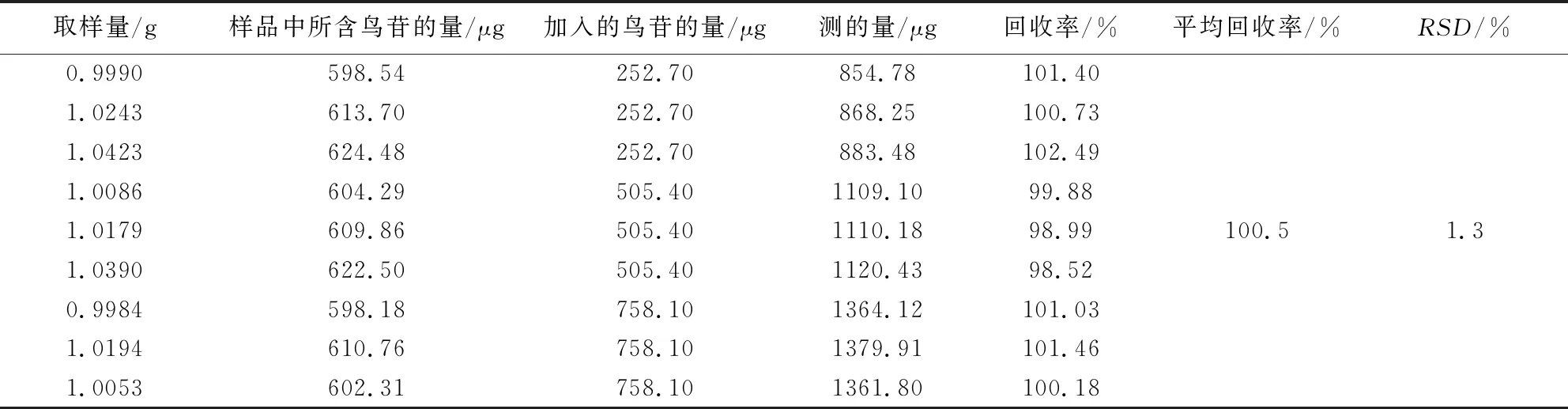
表3 板青颗粒中鸟苷加样回收率
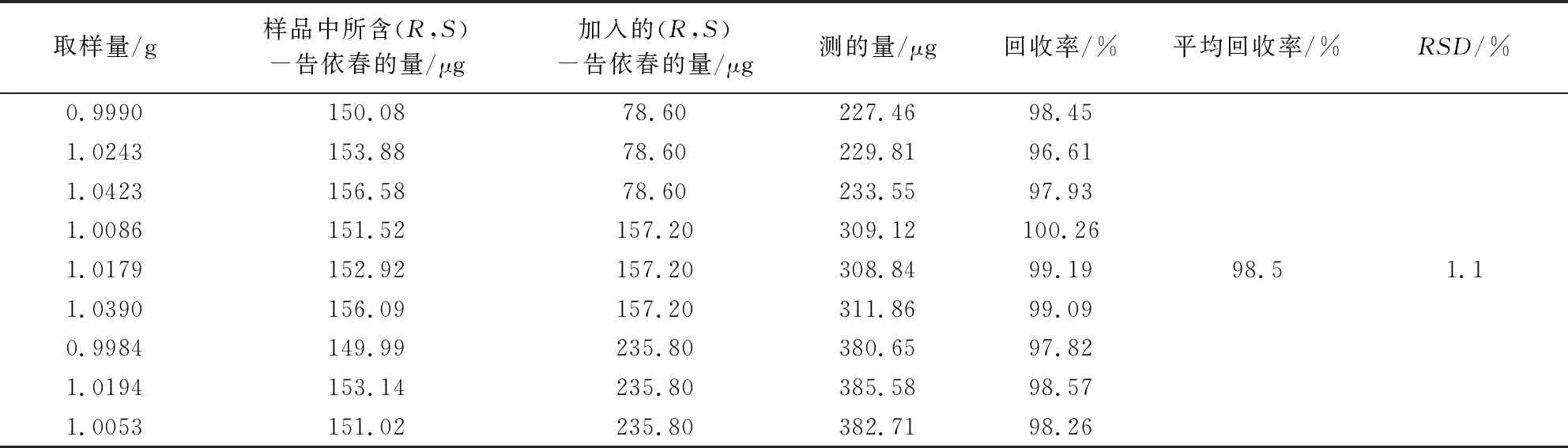
表4 板青颗粒中(R,S)-告依春加样回收率
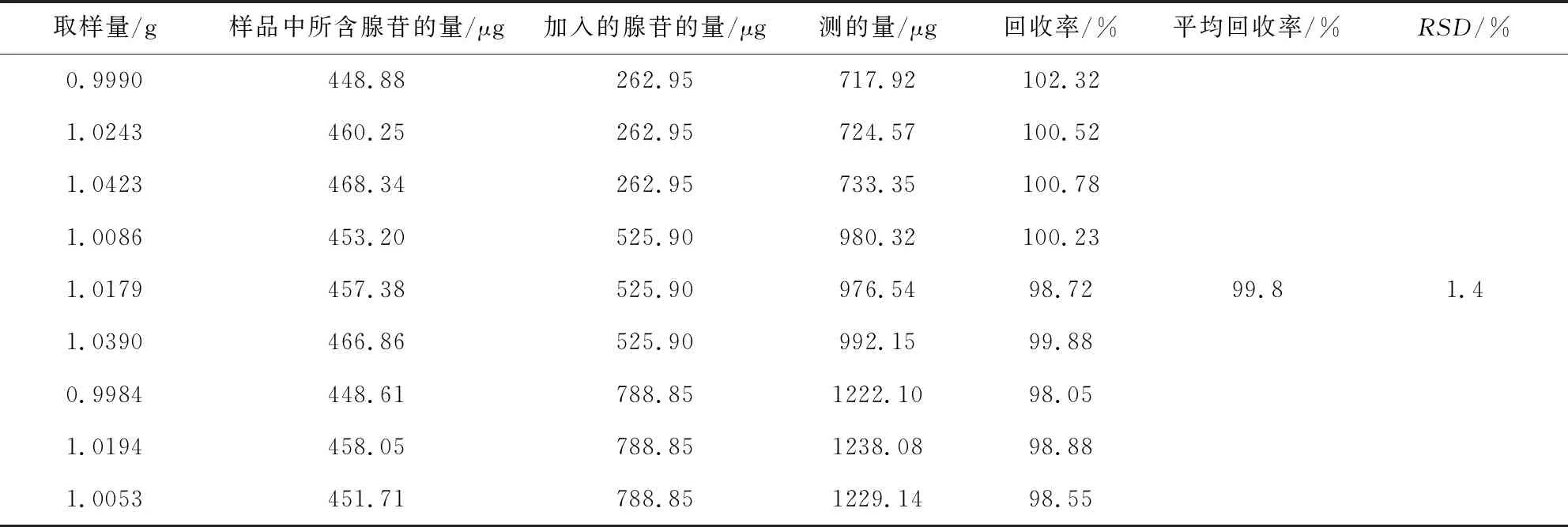
表5 板青颗粒中腺苷加样回收率
2.2.10 含量测定 取研磨后的板青颗粒细粉约2 g,精密称定,按“2.2.3”项下操作,制成供试品溶液。分别精密吸取混合对照品溶液与供试品溶液各10 μL,注入液相色谱仪,测定,按外标法以峰面积计算供试品中尿苷、鸟苷、(R,S)-告依春和腺苷的量,结果见表6。
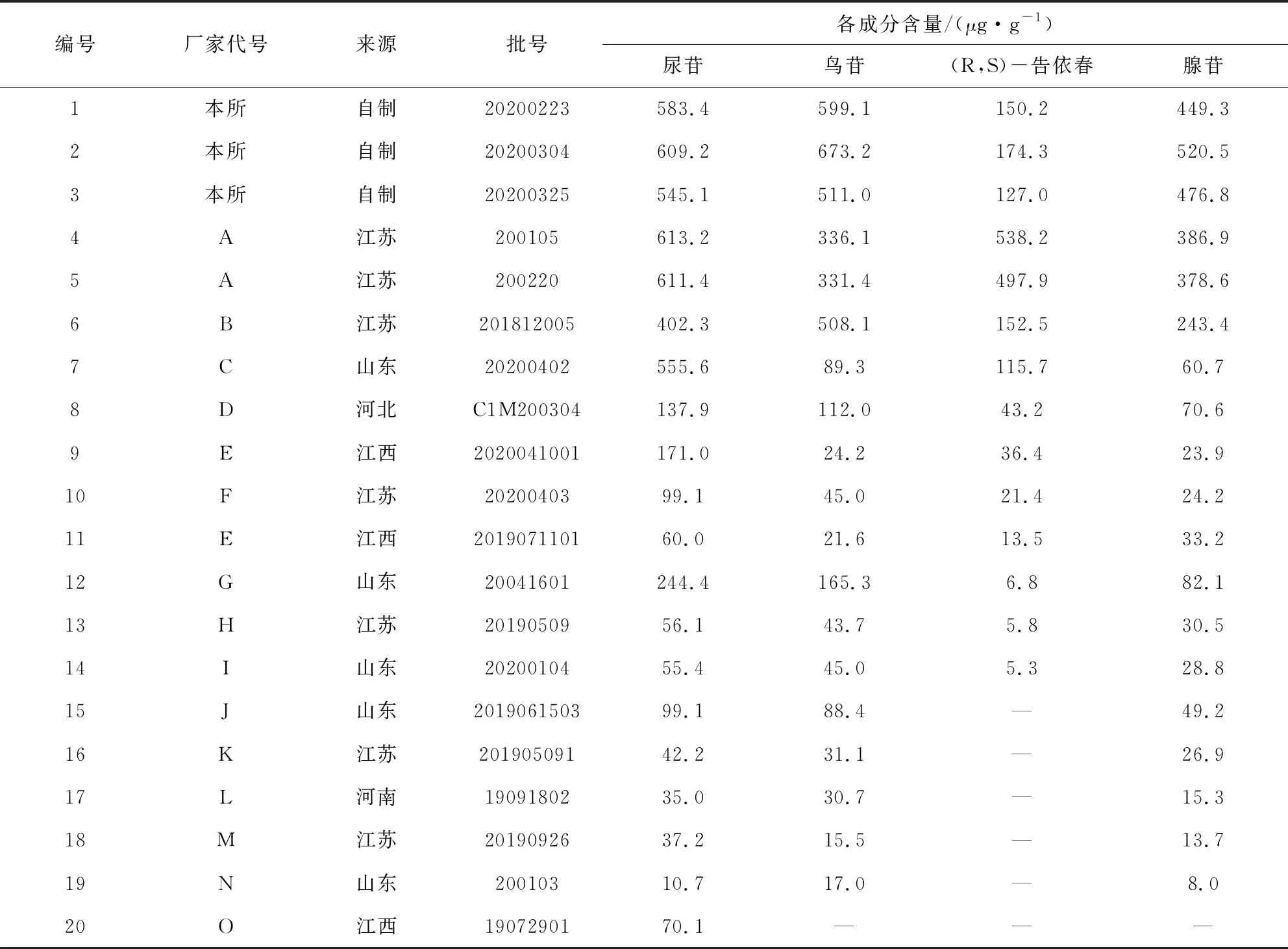
表6 板青颗粒4种成分含量测定结果(n=3)
3 讨论与结论
3.1 指标性成分的选择 方中板蓝根为十字花科菘蓝IsatisindigoticaFort.的干燥根,主要含有生物碱类、核苷类、氨基酸类等化学成分[10-12]。《中国兽药典》2015年版二部中板蓝根质量标准选择精氨酸进行薄层色谱鉴别,以(R,S)-告依春为指标成分进行薄层色谱鉴别和高效液相色谱定量测定,可实现与南板蓝根的快速鉴别。大青叶与板蓝根同出一源,为菘蓝的干燥叶,与板蓝根所含化学成分基本一致[13-14],但不同化学成分在两者中含量差距较大。大青叶现行质量标准以靛蓝、靛玉红为指标成分进行薄层色谱鉴别,并采用高效液相色谱法对靛玉红进行定量测定。本研究结合板青颗粒的临床功效,建立以靛玉红为代表的吲哚类成分薄层色谱鉴别方法;建立以(R,S)-告依春为代表的生物碱及以尿苷、鸟苷和腺苷为代表的核苷类成分的高效液相色谱定性定量测定方法。上述几种化合物在热水中溶解性好,用水煎煮提取时能够很好的溶出,且具有较强的抗菌、抗病毒活性[15-16],故可作为指标性成分来控制该制剂的质量。
3.2 精氨酸薄层鉴别的改进 按照现行标准的鉴别2进行精氨酸薄层鉴别时,经常会出现供试品溶液的精氨酸斑点相比精氨酸对照品的斑点滞后的现象,严重影响结果的判定。杨新等研究表明[17],板青颗粒的辅料蔗糖易溶于提取液稀乙醇,后在加热蒸干过程中被水解成还原糖,还原糖会进一步与供试品中的氨基酸或蛋白质发生美拉德反应(Maillard Reaction),形成大分子物质类黑精,导致薄层板上斑点滞后。本研究利用蔗糖在乙醇中溶解度很低的特性,采用乙醇做提取液,用自然挥发代替加热蒸干进行供试品溶液的制备,减少了供试品溶液中蔗糖的含量及还原糖的产生,有效解决了斑点滞后问题。
3.3 HPLC法定性及定量实验条件的选择 参考文献考察多种流动相,选择甲醇和水梯度洗脱,各峰分离度好。比较了Shim-pack VP-ODS柱,Capcell Pak C18 MG柱、Waters Atlantis T3柱等,样品溶液中(R,S)-告依春、尿苷、鸟苷和腺苷均可达到基线分离,选择245 nm作为检测波长时,各被测色谱峰均有较好的响应值。本研究预实验表明4种被测组分可使用不同的提取方法及展开条件进行TLC鉴别,但考虑到HPLC测定含量时,一针进样可同时检测4种成分,使用PDA检测器采集光谱图,将保留时间及光谱匹配度作为判定指标,亦可对4种成分进行定性鉴别,相比薄层色谱法前处理简单,受其他因素影响小。
3.4 评价性研究结果分析及建议 将自制样品及市场收集的17批样品按现行标准检测,均符合规定。用新建立的方法进行检测,20批样品中仅有14批能够进行(R,S)-告依春定量测定,其余6批均未测得或低于检测限,含量范围在0~538.2 μg·g-1;尿苷的含有量范围在10.7~613.2μg·g-1;鸟苷的含有量范围在0~673.2 μg·g-1;腺苷的含有量范围在0~520.5 μg·g-1。结果表明不同生产企业,同一生产企业的不同批次之间,4种成分含量差异很大。分析原因,首先是不同产地药材本身的差异性[18-21],药材中有效成分的含量直接影响制剂的质量。殷世宁等对不同产地的18批次板蓝根中尿苷、腺苷、(R,S)-告依春3种成分进行测定,(R,S)-告依春为0.034~0.347%,腺苷为0.001~0.079%,尿苷为0.010~0.051%;其次是否按照处方足量投料也直接关系成品含量;最后不同生产企业使用的提取工艺或制剂制法的差异,亦导致某些成分转移率较低[22]。参考《中国兽药典》中药品质量标准分析方法验证指导原则,含有量限度按照20批样品含量测定结果(表6)的平均值下浮20%计算,建议规定本品每1 g含尿苷、鸟苷和腺苷的总量不得少于0.46 mg;含(R,S)-告依春不得少于0.075 mg。
中兽药通过“多成分、多作用、多靶点”在我国兽医医疗系统中发挥了重要作用,且不易产生耐药性,是国家实施兽用抗菌药使用减量化行动可行的抗菌药替代方法之一,其主成分含量的差异会直接导致临床疗效的优劣。板青颗粒作为临床常用中兽药,疗效确切,但其质量控制水平还比较落后,不利于日常监管。综上所述,建议《中国兽药典》2020版完善板青颗粒的鉴别项、增加含量测定项,为有效控制产品质量提供技术支持。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
检察风云(2022年5期)2022-04-05
中医眼耳鼻喉杂志(2021年2期)2021-07-21
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
临床医药文献杂志(电子版)(2017年11期)2017-05-17
国外医药(抗生素分册)(2016年3期)2016-07-12
海峡科技与产业(2016年3期)2016-05-17
云南中医学院学报(2015年3期)2015-07-31
中国合理用药探索(2014年11期)2014-03-11
