
乳腺癌关键基因的生物信息学分析
2021-03-06 01:40李淑怡黄玉珍蓝秀万
广州医药 2021年1期
李淑怡 黄玉珍 蓝秀万
广西医科大学基础医学院(南宁 530021)
乳腺癌是全球第二大常见癌症,也是我国第二位最常见的恶性肿瘤[1]。乳腺癌是一种复杂的、表型多样性的疾病,发病机制仍有很多未知,迫切需要更多的遗传信息来提供诊断和治疗参考。此外,基于Bretthauer M[2]的研究表明,提早发现恶性肿瘤从而改变可控的致癌因素可以通过早期筛查来实现,普及早期筛查可以降低患者死亡率。因此,寻找乳腺癌新的生物标志物和治疗靶点已成为趋势。近年来,各国学者发布大量癌症高通量测序数据,生物信息学方法能够克服以上测序数据由于不同技术平台或小样本分析带来的局限性,从而发现了大量有价值的生物信息[3]。本研究旨在从生物信息学角度识别与乳腺癌发病和预后相关的关键基因,为其筛查,诊断和治疗提供新的候选靶点。
1 材料与方法
1.1 数据获取
基因综合表达数据库(GEO,http://www.ncbi.nlm.nih.gov/geo)是一个公共的功能基因组学数据存储库,包含了大量的芯片、微阵列和基因表达数据。我们从GEO数据库中下载了三个基因表达数据集(GSE36295、GSE71053、GSE86374)。根据平台中的注释信息,将探针转化为相应的基因符号。GSE36295数据集包含45例乳腺癌组织样本和5例非癌组织样本。GSE71053包含6例乳腺癌样本和12例非癌样本。GSE86374包含124例乳腺癌样本和35例非癌样本。
1.2 差异基因的筛选
利用基于R语言Biobase, GEOquery, limma工具包运作的GEO2R(http://www.ncbi.nlm.nih.gov/geo/geo2r)筛选乳腺癌与非癌组织间的差异基因(differentially expressed genes,DEGs)。GEO2R是一个交互式的在线工具,它可以用来比较GEO中的两个或多个数据集,以便识别差异基因。校正后的P值(adj.P)和Benjamini和Hochberg伪发现率可以为发现有统计学意义的基因和限制假阳性之间提供平衡,移除不具有相应基因符号的探针集并将多探针集对应的基因数据取平均值。|log2FC(fold change)|≥1、adj.P值<0.01具有统计学意义。共筛选出95个DEGs,使用bioin-formatics(http://bioinformatics.psb.ugent.be/webtools/Venn/)在线数据库进行DEGs的维恩图制作。
1.3 PPI网络构建与模块分析
利用互作基因检索工具STRING数据库(http://string-db.org)构建蛋白质相互作用网络(PPI),之后使用Cytoscape(3.8.0版本)进一步绘制PPI网络。Cytoscape是一个用于可视化分子相互作用网络的开源生物信息学软件平台。使用软件中基于最大团中心性(maximal clique centrality, MCC)算法的cytoHubba插件对所有DEGs进行筛选,并根据Degree进行排序,筛选出排名前十的基因作为核心(Hub)基因。MCODE是一款基于拓扑分析对给定网络进行聚类的插件,使用MCODE识别PPI网络中最重要的模块。选择标准:MCODE评分>10,degree cut=2, node score cut=0.2, Max depth=100, k-score=2。
1.4 基因本体(GO)和京都基因和基因组百科全书(KEGG)富集分析
DAVID数据库(The Database for Annotation, Visualization and Integrated Discovery, https://david.ncifcrf.gov/)是一个集生物数据和分析工具于一体的在线生物信息库,GO可对基因进行注释及富集分析,以研究基因的生物功能,包括生物过程(BP)、分子功能(MF)和细胞成分(CC)。KEGG是一个数据库资源,用于高通量实验中获取的大规模分子数据,集中理解其高级功能和生物系统,可用于通路分析。使用DAVID对95个DEGs和10个Hub基因进行KEGG和GO分析。P<0.05存在显著性差异。
1.5 Hub基因的验证
用GEPIA(http://GEPIA.cancer-pku.cn/)对Hub基因进行进一步验证与生存分析。GEPIA是基于癌症基因组图谱(TCGA)和基因型组织表达(GTEx)数据库的网站工具,通过对肿瘤和正常数据进行基因表达分析,将结果绘制成箱线图。应用生存曲线库(K-Mplotter,https://kmplot.com/)分析肿瘤中Hub基因与死亡时间的关系。利用在线数据库Oncomine(https://www.oncomine.org/)对某些基因在不同肿瘤类型中的表达情况以及在乳腺癌各个研究中的结果进行分析。
2 结 果
2.1 差异基因
在GEO数据库中选取的3个(GSE36295、GSE71053、GSE86374)包含乳腺癌及正常肺组织基因芯片的数据集共计227例样本被纳入本项研究,并对其中175例乳腺癌和52例正常肺组织进行了分析,共筛选出95个DEGs(62个上调基因及33个上调基因),见图1。
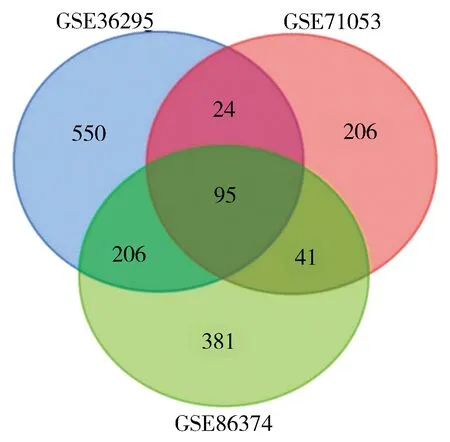
图1 维恩图
2.2 PPI网络及Hub基因筛选
在STARING数据库基础上,通过Cytoscape 软件构建 PPI 网络(图2)。应用cytoHubba模块筛选出分值最高的前十个基因作为Hub基因:CENPF、KIF2C、TOP2A、NUSAP1、HMMR、MELK、KIF4A、ASPM、CEP55、CCNB1。应用MCODE插件共检测到2个评分>10的模块,将第一个模块作为最显著模块(图3),最显著模块中共有46个节点和1 007条连线。
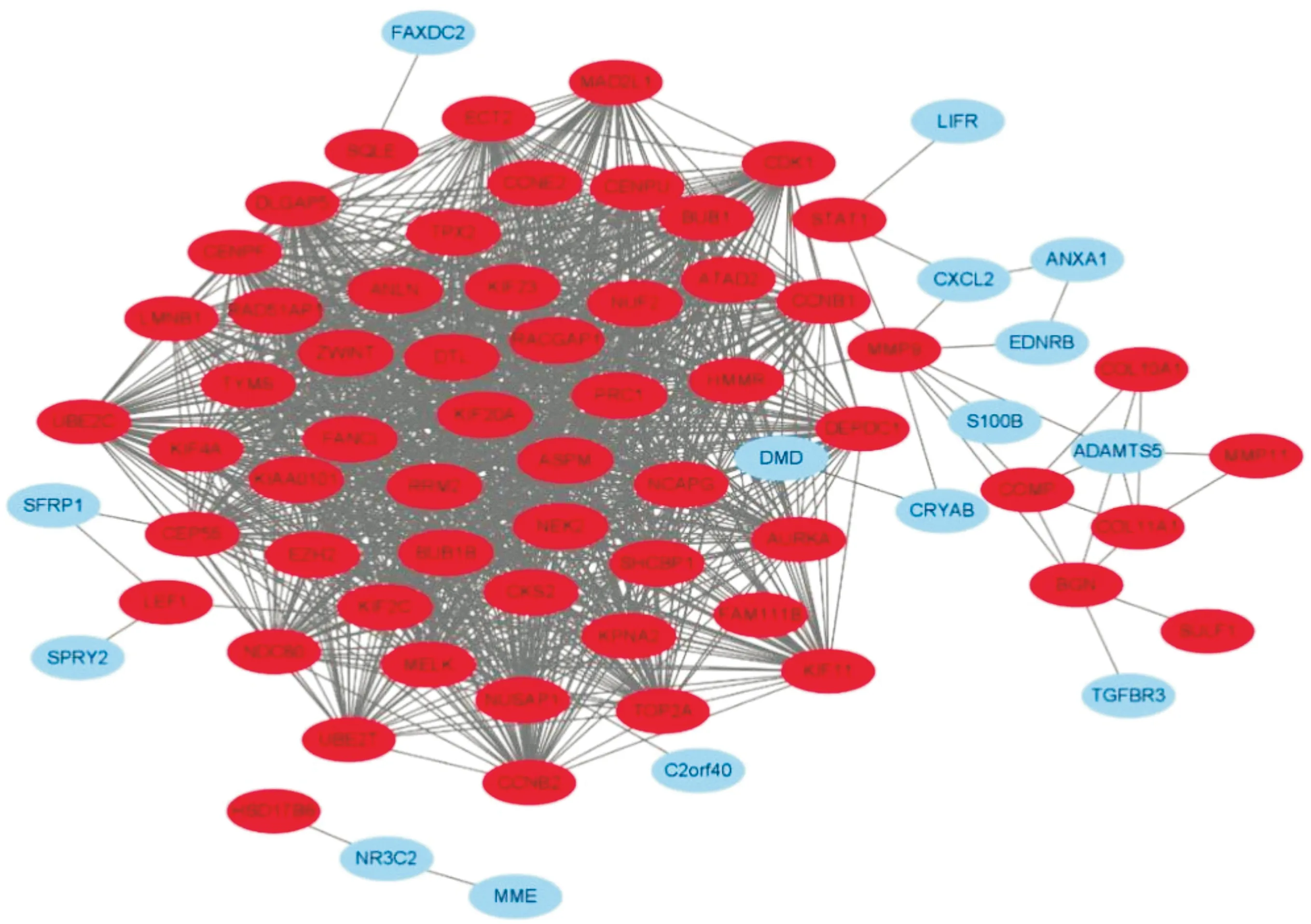
图2 PPI网络结构图,红色为上调基因,蓝色为下调基因
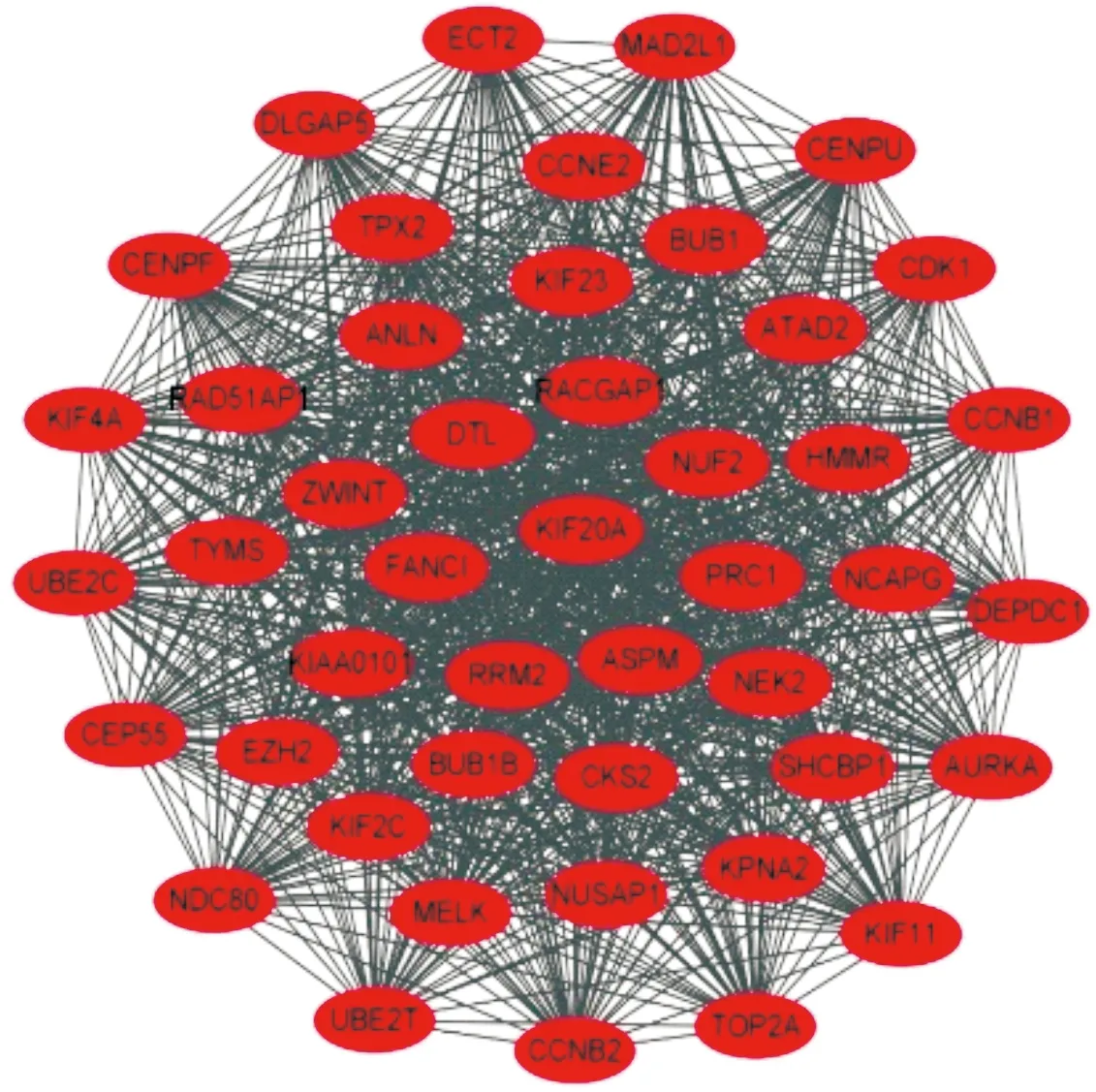
图3 最显著模块
2.3 DEGs与Hubgenes的生物学注释
如图4所示,为了进一步了解差异基因的功能,应用DAVID数据库进行GO与KEGG富集分析,以P<0.05为筛选条件,选取富集显著性排序前5的GO和KEGG通路作图。结果表明:仅上调的DEGS有统计学意义,上调的DEGs的GO生物学过程(BP)主要富集在M期、有丝分裂、核分裂、细胞器分裂等。GO细胞组分(CC)主要富集在纺锤体、微管、细胞骨架、着丝粒等。GO分子功能(MF)主要富集在腺苷酸结合、嘌呤核苷结合、微管运动活性、ATP结合等。Hub基因全部为上调基因,GO富集分析结果如表1,主要集中在有丝分裂、细胞周期和无膜细胞器等过程。
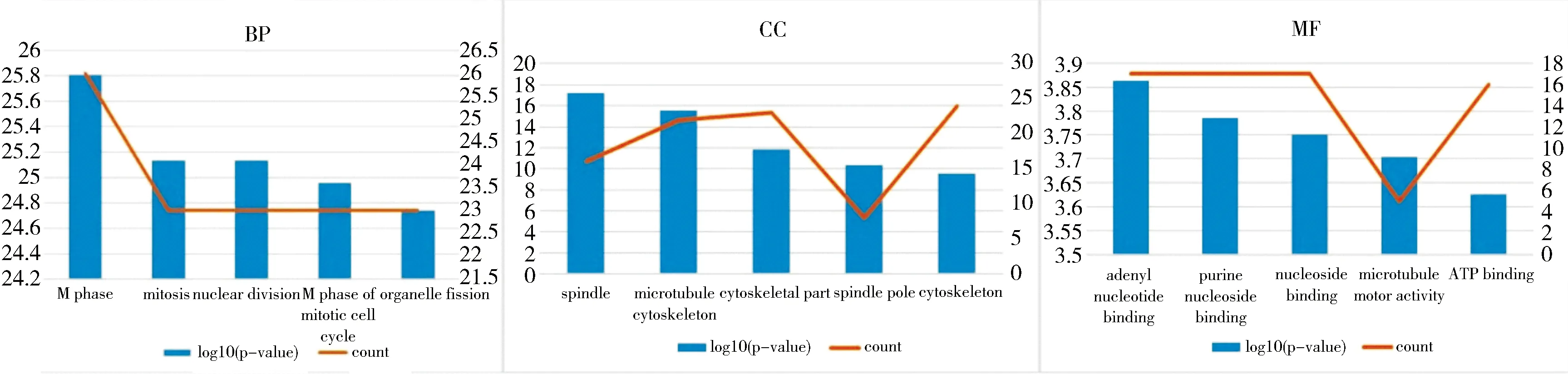
图4 DEGs的GO富集分析
2.4 DEGs的KEGG信号通路
DEGs的KEGG通路富集有四条:卵母细胞减数分裂、细胞周期、p53信号通路和孕酮介导的卵母细胞成熟通路。结果如图5。

图5 DEGs的KEGG富集分析
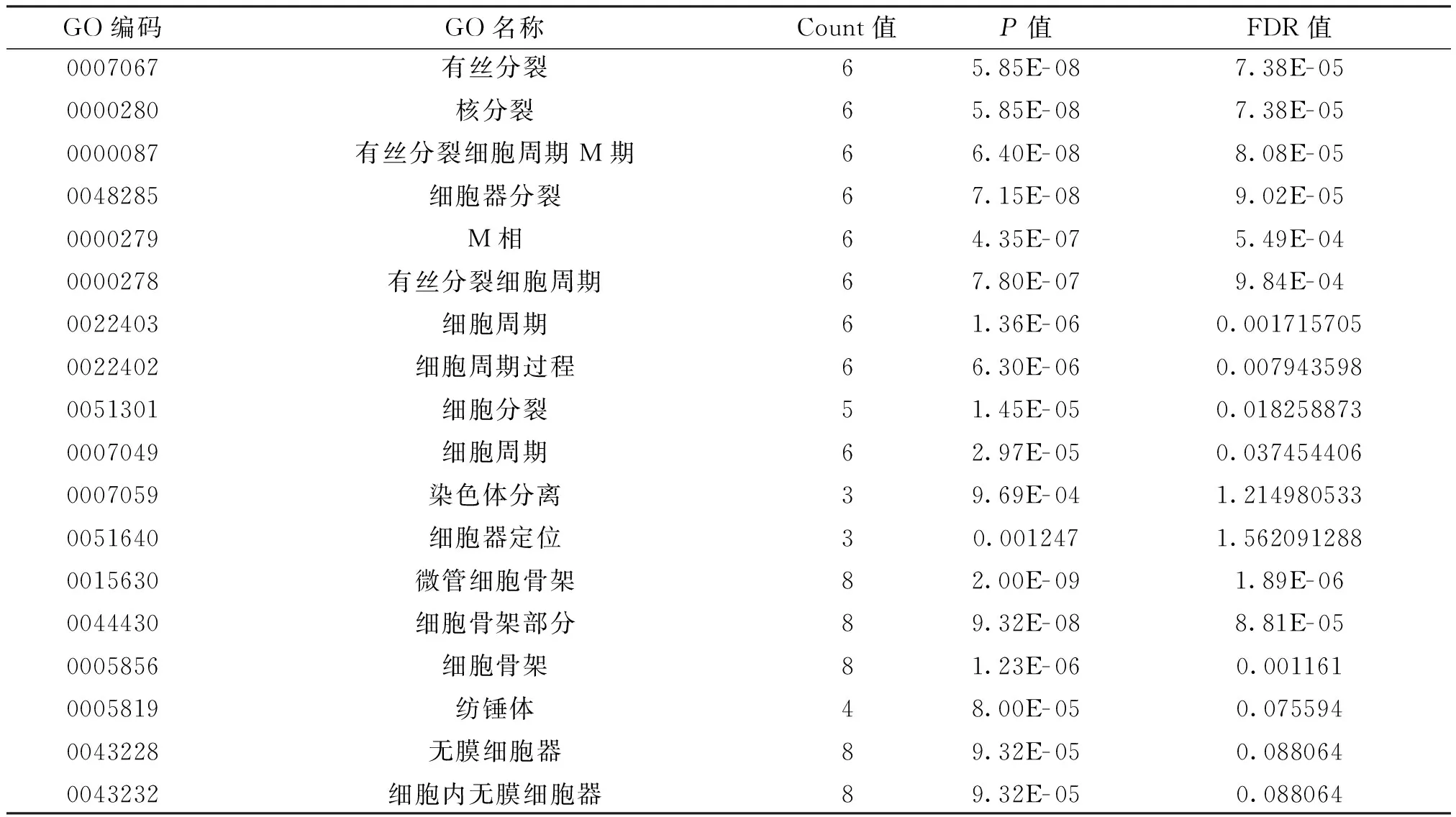
表1 Hub基因的GO富集分析
2.5 数据验证
通过GEPIA数据库分析,相较正常组织,CENPF、KIF2C、TOP2A、NUSAP1、HMMR、MELK、KIF4A、ASPM、CEP55、CCNB1均在乳腺癌中高表达(图6)。
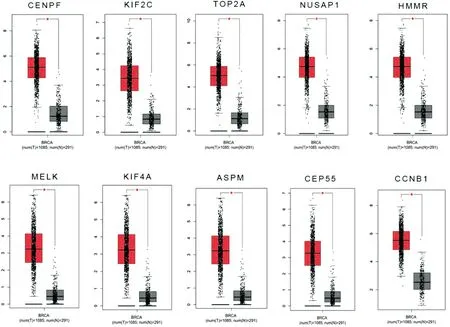
图6 Hub基因在乳腺癌中的表达情况
2.6 CENPF与KIF2C在各类癌种中的表达情况
采用Oncomine数据库分析cytoscape插件计算得到的Hub基因中分值最高的两个基因:CENPF与KIF2C在各癌种中的表达情况,其中97项研究提示CNEPF表达增高,9项提示表达降低。85项研究提示KIF2C表达增高,10项提示表达降低(图7)。

图7 CENPF与KIF2C在多类癌种中的表达差异情况
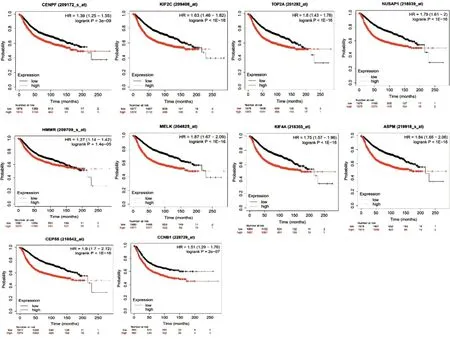
图9 Hub基因表达量对乳腺癌患者预后影响
2.7 CENPF与KIF2C在乳腺癌中的表达情况
通过Oncomine中Curtis[4]和Xiao-Jun Ma[5]的研究成果进行Meta分析,结果提示:与正常组织相比,CENPF与KIF2C均在乳腺癌组织中表达量升高,差异有统计学意义(P<0.05),见图8。

图8 CENPF与KIF2C在乳腺癌中的总体表达情况
2.8 Hub基因与预后的关系
本研究采用K-M Plotter评价10个Hub基因的预后价值,分析结果(图9)显示,10个基因均与乳腺癌的总体生存率相关,差异有统计学意义(P<0.05);高表达组生存时间更短。
3 讨 论
在全球范围内,乳腺癌是女性最常见的肿瘤[6]。中国每年新发乳腺癌26.9万例,死亡7.0万例,中位年龄为50岁,同时乳腺癌也是导致45岁以下女性死亡的最常见原因[7]。本研究采用生物信息学方法为今后进一步探讨乳腺癌发生和发展的潜在分子机制提供靶点。本研究共筛选出95个差异基因,其中33个下调基因,62个上调基因,构建PPI网络,并结合GO和KEGG富集分析用于探索DEGs之间的相互作用。上调基因GO主要富集于M期、有丝分裂、纺锤体和ATP结合等过程中,KEGG主要集中在卵母细胞减数分裂、细胞周期、p53信号通路和孕酮介导的卵母细胞成熟通路中。有研究表明,在肿瘤的发生、发展中,细胞周期过程和有丝分裂细胞周期的失调起着重要作用[8]。这与我们的结果是一致的。
通过Cytoscape软件筛选出10个过表达Hub基因 :CENPF、KIF2C、TOP2A、NUSAP1、HMMR、MELK、KIF4A、ASPM、CEP55、CCNB1。其中CENPF和KIF2C是通过Cytoscape插件计算分值最高的两个基因。CENPF是一种细胞周期相关的核抗原,在G0/G1细胞中低水平表达,在S期积聚在核基质中,在G2/M细胞中表达量最大。有研究表明,CENPF在多种人类恶性肿瘤中被鉴定为细胞增殖的标志物,且其高表达有助于肝癌细胞的增殖[9]。
KIF2C是有丝分裂着丝粒相关的运动蛋白。它参与微管分解、双极纺锤体形成和染色体分离,以调节有丝分裂和细胞周期,对正常纺锤体组装并修复微管和染色体异常具有重要意义[10]。因此,在理论上,KIF2C的异常表达可能在肿瘤的发生发展中起重要作用。有研究表明KIF2C蛋白在舌癌组织中的表达明显高于癌旁组织,且KIF2C的高表达与淋巴结转移和肿瘤分期有关[11]。结合我们研究发现KIF2C高表达患者生存时间更短(图9)。KIF2C的过度表达极有可能促进乳腺癌的发展。
TOP2A位于人表皮生长因子受体2(HER2)扩增子下游的一个单独的扩增子中。研究发现TOP2A拷贝数的改变与激素受体(HR)和HER2状态密切相关,但TOP2A作为预后指标的价值可能有限[12]。NUSAP1(核仁和纺锤体相关蛋白1)通过促进微管的聚集来控制细胞周期,在纺锤体的组装和形成中起着重要作用。Xi Zhang等人[13]发现NUSAP1表达下调可以抑制浸润性细胞癌的增殖、迁移和侵袭,增加癌细胞对阿霉素(E-ADM)的敏感性。Huizhong Zhang等人[14]证明HMMR(透明质酸介导的运动受体)激活TGF-b/Smad2信号诱导上皮-间质转化通路,揭示了HMMR在胃癌化疗耐药中的重要作用,并提示HMMR可能是该病的潜在预后标志物或治疗靶点。MELK被认为是多种癌症类型的癌症依赖和潜在的药物靶点,在这些癌症中过表达,并且MELK的高表达与患者预后不良有关[15]。KIF4A在多种细胞过程中起重要作用,主要包括染色体凝聚和分离、中纺锤体形成和有丝分裂过程中的胞质分裂。KIF4A在多类肿瘤的发生发展中起着重要的作用,有实验推测KIF4A可能是乳腺癌强有力的预后预测因子,且靶向KIF4A活性可能是乳腺癌治疗的一个有前景的治疗选择[16]。CEP55已被证明在多种癌细胞系模型中调节细胞增殖、迁移和侵袭。Jeffery J等人[17]发现CEP55是丝裂原活化蛋白激酶(MAPK)MYC信号传导的下游效应器。CCNB1(又称CyclinB1)属于高度保守的细胞周期蛋白家族,在各种癌症中都有显著的高表达。有报道称,CCNB1可能参与上皮-间质转化(EMT)和转移的过程[18]。
总之,本研究旨在通过整合多个基因芯片数据筛选了可能参与肝癌的发生或发展的95个DEGs和10个Hub基因,可作为乳腺癌的诊断和预后标志物。但这些基因在乳腺癌中的生物学功能尚需进一步研究。
猜你喜欢
中国医学科学院学报(2022年1期)2022-12-30
诊断学(理论与实践)(2020年2期)2020-06-28
心电与循环(2020年1期)2020-02-27
国际呼吸杂志(2019年4期)2019-03-12
生物学教学(2017年9期)2017-08-20
江苏农业科学(2017年5期)2017-04-15
中华老年多器官疾病杂志(2016年7期)2016-04-28
癌症进展(2016年10期)2016-03-20
医学研究杂志(2015年5期)2015-06-10
湖北农业科学(2014年3期)2014-07-21
