
超高效液相色谱-高分辨质谱测定10种植物内源激素
2021-03-05 09:40王安琦卢亚萍
分析科学学报 2021年1期
汪 瑾, 戴 琳, 王安琦, 卢亚萍*
(南京农业大学生命科学学院生命科学实验中心,江苏南京 210095)
植物激素是一类小分子化合物,参与植物的抗生物和非生物胁迫[1,2]。植物激素通常分为六大类,包括生长素、赤霉素、细胞分裂素、脱落酸、乙烯和油菜素甾醇[3]。植物激素一般是极低浓度的植物内源物质,需要使用合适的方法来提取富集。用于分析植物激素的样品制备过程,包括采样、提取、纯化和浓缩,己成为快速分离以及痕量植物激素超灵敏分析的一个瓶颈[4]。
植物组织中植物激素的浓度和分布受温度、水情、空气湿度和光照强度等条件的影响[5]。因此,从植物中分离组织后,需要立即将收集的新鲜组织冻结在液氮中。然后选取合适的提取溶剂,理想的提取溶剂能够高效率地从样品中提取尽量多种类的植物激素。用于激素提取的溶剂包括甲醇、乙腈、丙酮、异丙醇等[6]。样品前处理对实验结果产生很大的影响[7]。前处理技术,包括液-液萃取、固相萃取、分子印迹萃取、免疫亲和纯化等,现已被用于从粗提物中进一步纯化痕量植物激素[8]。检测技术一般包括生物测定法、免疫测定法、电分析法、色谱法以及质谱法[9-12]。色谱和质谱联用技术具有强分离能力,高灵敏性、选择性和准确性,已成为激素测定的强有力的工具。因此,液相色谱-四极杆飞行时间-质谱(LC-qTOF-MS)方法现已被用于测定痕量植物激素[13-15]。
本研究的目的在于建立一种利用LC-qTOF-MS测定植物中常见的内源激素(赤霉素、脱落酸、细胞分裂素、茉莉酸、水杨酸)的快速高效的方法,为激素的功能、相互作用等研究奠定基础。
1 实验部分
1.1 仪器与试剂
Waters G2-XS Q-TOF质谱仪(美国,Waters公司),配有电喷雾(ESI)离子源;SpeedVac真空离心浓缩仪(美国,Thermo Fisher公司)。
10种植物激素标准品(纯度均>97%):脱落酸(ABA)、赤霉酸(GA)、茉莉酸(JA)、水杨酸(SA)、茉莉酸甲酯(MJ)、吲哚乙酸(IAA)、异戊烯腺嘌呤(IP)、玉米素(ZT)、玉米素核苷(ZR)、独脚金内酯(SL),均购自上海源叶生物科技有限公司。将10种标准品用甲醇溶解,配制成浓度为1 mg/mL的母液,使用时稀释。甲醇、乙腈、丙酮、甲酸和NH4Ac为色谱纯,购自Merck Millipore公司;其他试剂为国产分析纯。
小麦、水稻、烟草、棉花、番茄、拟南芥来自江苏农科院种质资源与生物技术研究所,温室培养。
1.2 样品提取
取幼苗期的各种植物材料,用液氮研磨破碎后,称取100 mg,分别加入1 mL不同的样品提取液(1:66.7%异丙醇+1%甲酸;2:80%丙酮+1%甲酸;3:80%甲醇+1%甲酸;4:乙腈;5:乙腈+1%甲酸),超声提取60 min,13 000 r/min离心10 min,收集上清液。沉淀中再加入0.5 mL样品提取液振荡10 min,13 000 r/min离心10 min,收集上清液。合并两次上清液,用真空离心浓缩仪挥干后,重新溶解于含0.1%甲酸甲醇中。
1.3 固相萃取
用1 mL 纯甲醇淋洗固相萃取小柱两次,再用1 mL 0.1%甲酸淋洗柱子两次。将10%甲醇(含0.1% 甲酸)溶解的样品加入到柱子里,用1 mL 0.1%甲酸淋洗柱子,最后用1 mL 80%甲醇(含0.1% 甲酸)洗脱两次。洗脱后的组分用真空离心浓缩仪挥干,复溶于0.2 mL 80%甲醇(含0.1% 甲酸)中,待测。
1.4 液相色谱-质谱检测条件
色谱条件:色谱柱:UPLC BEH C18柱(100×2.1 mm,1.7 μm;美国Waters公司);流动相A为0.1%甲酸+2 mmol/L NH4Ac(正离子),或2 mmol/L NH4Ac(负离子);流动相B为甲醇。洗脱条件如下:0~11 min,5%~95%B;95%B淋洗柱子,然后恢复到起始组分5%B。进样量2 μL;柱温35 ℃。
质谱条件:毛细管电压:3.0 kV;锥孔电压:40 V;源温度:120 ℃;脱溶剂温度:400 ℃;碰撞能量:10~50 eV;比扫描范围:m/z50~1 000;亮氨酸脑啡肽(LE)用于质量校正。
1.5 数据分析
质谱数据采集和分析使用 Waters Masslynx 4.1软件。实验数据为三次重复平均值,表示为平均数值±标准偏差(SD)。采用StudentT检验对实验数据进行比较,当p<0.05时,认为有显著性差异;当p<0.01时,认为差异极为显著;当p>0.05时认为没有显著性差异。
2 结果与讨论
2.1 色谱条件的选择及优化
色谱柱及流动相的选择会对化合物的分离度和峰形产生重要的影响[19,20]。液相色谱-质谱分析中,有机流动相一般采用乙腈或甲醇,同时也会添加一定量的酸和盐以提高离子化效率、改善峰形。本研究比较了乙腈和甲醇的分离效果,发现两者没有显著差异,于是选择了毒性较小、成本较低的甲醇作为有机相。同时在正离子模式下,流动相A(ddH2O)中添加了0.1% 的甲酸及2 mmol/L的NH4Ac,负离子模式下流动相A中添加了2 mmol/L的NH4Ac,达到了良好的离子化效率和分离效果。
2.2 10种常见植物内源激素的定性分析
在MS/MS采集模式下,对10种化合物进行一级全扫描和二级碎片检测。正离子模式下,IAA、IP、ZR、ZT、SL和MJ有较好的响应度,而ABA、GA3、JA和SA在负离子模式下响应度较好,提取离子色谱图如图1、图2。在MS/MS 模式下,以[M+H]+或[M-H]-离子为母离子得到二级碎片离子的质谱信息列于表1。选择丰度相对较高的子离子作为特征碎片离子,各物质的一级、二级色谱图、质谱图及结构信息如图3、图4。
2.3 10种植物内源激素的定量测定
将10种激素标准品溶液稀释为0.1、0.2、0.5、1.0、2.0、5.0、10.0、20.0、50.0、100.0 ng/mL,对每个梯度的样品进行质谱测定,采集模式为MS、MS/MS或MRM。采用不同的采集模式时,各种激素的响应度不同,其中MS的响应度最高,而MS/MS及MRM模式下的响应度会降低1~2个数量级。因此采用MS模式对各种激素进行定量测定,制作标准曲线,确定其检测限(LOD)和定量限(LOQ),测定结果列于表2。所有的激素样品在稀释浓度范围内具有较好的线性关系,相关系数R2达到0.99以上。每种样品检测限和定量限相差很大,其中IP,ZR定量限为0.1 ng/mL,是所测激素中质谱响应度最高的。

图1 正离子模式下6种植物激素的提取离子色谱图Fig.1 Extraction ion chromatograms of six plant hormones in positive ion mode

图2 负离子模式下4种植物激素的提取离子色谱图Fig.2 Extraction ion chromatograms of four plant hormones in negative ion mode

图3 正离子模式下6种植物激素的一级、二级质谱图及碎片结构信息Fig.3 MS,MS/MS and structure elucidation of six plant hormones in positive ion mode

表1 10种植物激素的质谱信息

图4 负离子模式下4种植物激素的一级、二级质谱图及碎片结构信息Fig.4 MS,MS/MS and structure elucidation of four plant hormones in negative ion mode
2.4 10种植物激素添加回收实验
选用目标植物激素含量低于检测限的植物样品作为空白基质,分别加入3个浓度(10、50、200 ng/mL) 的激素标品混合物,通过Waters HLB固相萃取小柱回收后,测定回收前后的质谱响应值,计算测定浓度及回收率,测定结果见表3。对于浓度为200、50 ng/mL激素样品,回收率可达到90%以上;而对于浓度为10 ng/mL 的样品,回收率偏低。其中SL的回收率最低,为77.8%,ZT的回收率最高,为92.6%。
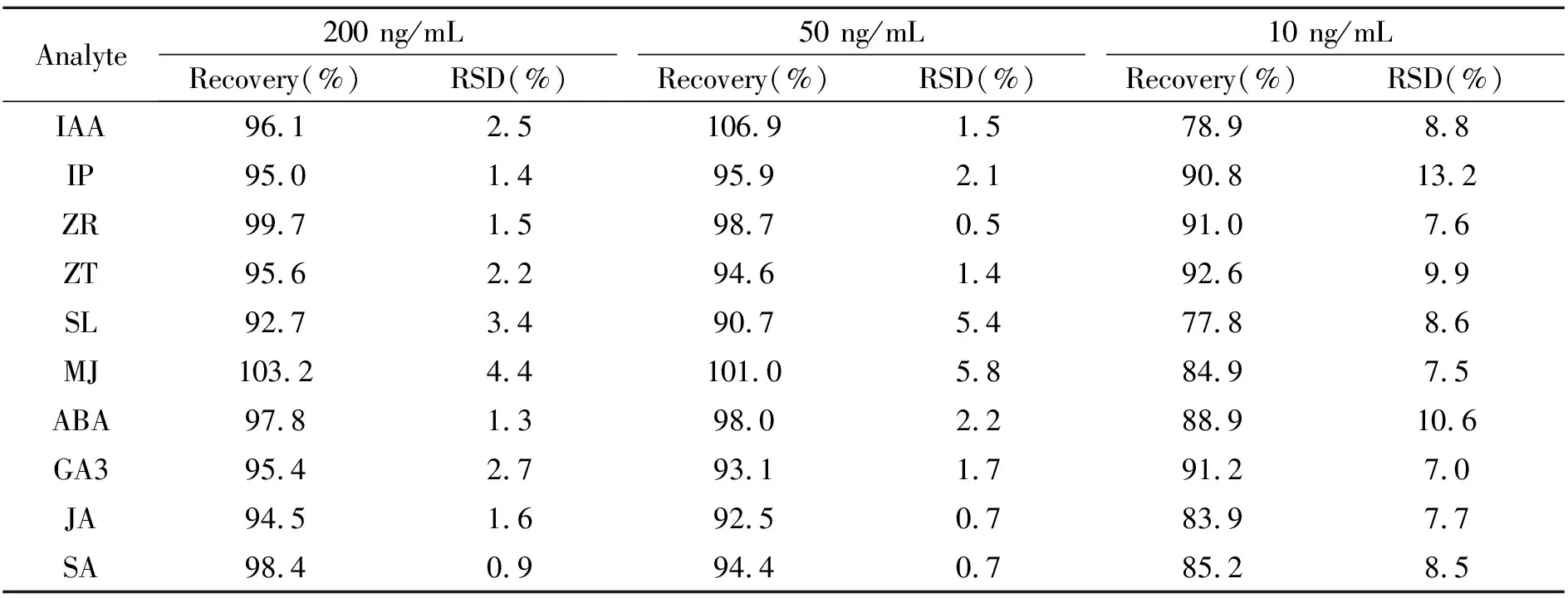
表3 10种植物激素的添加回收率和相对标准偏差(n=3)
2.5 不同的样品提取液和溶剂对激素提取效率的影响
选取5种试剂从烟草幼苗中提取激素。5种提取液分别是:(1)66.7%异丙醇+1%甲酸;(2)80%丙酮+1%甲酸;(3)80%甲醇+1%甲酸;(4)乙腈;(5)乙腈+1%甲酸。不同提取液的提取效果如图5。丙酮作为提取剂,提取液颜色很深,说明其中溶解了很多的叶绿素,会对后续的样品前处理和样品测定造成较大影响。异丙醇和乙腈的提取效果相对更好,而乙腈提取时IP的响应值明显优于异丙醇。加入1%甲酸对乙腈的提取效果有更好的提高作用。对于烟草样品中含量较高的成分IP、 ZR、 ZT、 SA,使用乙腈+1%甲酸作为提取液,提取效果明显优于其余几种提取液。
为了考察不同的溶剂复溶对于激素溶解和测定的影响,将提取后的激素初提液旋转挥干后溶于20%~100%的甲醇中,通过LC-MS测定其质谱响应值,结果见表4。对于IP,ZT和SA,在80%甲醇中检测信号最强,而ZR在100%甲醇中检测到的信号最强。100%甲醇溶解时溶液中叶绿素含量较高。综合考虑,选用80%的甲醇作为溶剂复溶激素提取物。
在提取过程中,比较了振荡提取和超声提取的提取效果,以及提取时间对提取效果的影响。结果发现,超声提取的植物激素的量大于振荡提取,并且提取60 min可达最佳效果。在对样品粗提液进行浓缩干燥时,我们比较了使用氮吹仪和离心浓缩仪的效果。两种仪器的处理对于植物激素的影响程度没有显著区别,因此选择速度更快的真空离心浓缩仪作为处理仪器。
实验还比较了汉邦C18柱、安普C18柱和MAX(阴离子交换吸附柱)的纯化效果,两种C18柱的回收效果略低于Oasis HLB小柱,而MAX柱在本实验中对于酸性化合物的回收效率并未显著高于HLB小柱,因此选用HLB柱作为标准品和植物样品的萃取柱。
2.6 植物材料中激素的测定
选取几种植物材料幼苗,按优化的提取及测定方法,通过高分辨率飞行时间-质谱,采用一级全扫描的方式测定其质谱响应值,并根据标准曲线计算各种激素的含量。从100 mg水稻、小麦、烟草、棉花、番茄、拟南芥幼苗中,检测到了IAA、IP、ZR、ZT、ABA、GA3、JA、SA(表5),而SL和MJ含量低于定量限,未检出。

表5 几种植物材料中植物激素的定量结果(ng/g FW)
3 结论
本研究优化了从植物样品中提取和纯化激素,并利用超高效液相色谱-高分辨率质谱进行定性定量测定。10种激素在质谱上均有良好的响应,检测限为0.05~5 ng/mL,定量限为0.1~10 ng/mL。采用含1%甲酸的乙腈超声浸提小麦、水稻、烟草、棉花、番茄和拟南芥幼苗粉末,然后用固相萃取小柱进行样品前处理,最后用80%甲醇复溶,采用质谱全扫描模式测定各种植物激素的含量。本实验建立了简便快速提取和测定植物激素的分析方法。
猜你喜欢
中草药(2022年19期)2022-10-14
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
农产品加工(2022年4期)2022-03-11
科学与财富(2021年13期)2021-07-04
中国科技教育(2017年9期)2018-01-23
食品界(2017年12期)2018-01-20
湖北农业科学(2014年12期)2015-01-06
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
中国信息化·学术版(2013年3期)2013-06-25
