
SIRT1在心血管疾病中的研究进展
2021-02-16 06:43武丹祝德秋同济大学附属同济医院药剂科上海200065
中南药学 2021年12期
武丹,祝德秋(同济大学附属同济医院药剂科,上海 200065)
心血管疾病(CVD)是我国城乡居民的首要死亡原因,患病人数推测为2.9 亿,且患病率和死亡率仍处于上升阶段,因此对其防治刻不容缓[1]。组蛋白去乙酰化酶(HDAC)通过介导蛋白质去乙酰化调控蛋白质的功能或活性,根据与酵母中组蛋白的同源性,哺乳动物中HDACs 可以分为4 类:Ⅰ型包括HDAC1/2/3/8,仅存在于细胞核中;Ⅱ型包括HDAC4 ~7,9 ~10,可以在细胞核和细胞质中穿梭;Ⅲ型包括沉默信息调节因子2 相关酶(SIRT),包括7 个同源类似物SIRT1 ~7,活性依赖于烟酰胺腺嘌呤二核苷酸(NAD+);Ⅳ型包括HDAC11。研究表明,不同的HDACs 对于CVD 有着不同的调控作用,其中Ⅲ型HDAC SIRT1 可通过介导下游蛋白分子赖氨酸残基的去乙酰化,调控心力衰竭、心肌缺血再灌注损伤、心肌肥厚、糖尿病心肌病、动脉粥样硬化等疾病的发生发展;同时SIRT1 激动剂白藜芦醇也表现出了良好的心血管保护潜力。本文将就SIRT1 调控的信号通路在CVD 中的作用进展进行综述。
1 SIRT1 的特征
1987年,Rine 等[2]发现了一种能够使酵母交配型基因沉默的蛋白质,将其命名为沉默信息调节因子2(sir2)。在随后的研究中,人们相继在哺乳动物体内发现了7 种sir2 的同源基因,分别命名为SIRT1 ~7,统称为sirtuin 家族。哺乳动物的SIRT 具有不同的亚细胞定位:SIRT1 和SIRT2 存在于细胞核和细胞质,SIRT3、SIRT4 和SIRT5 存在于线粒体,SIRT6 和SIRT7 位于细胞核[3]。SIRT 是依赖于NAD+的HDAC。NAD+是SIRT 去乙酰化过程的限速酶,NAD+与NADH浓度的比值越高,SIRT 的去乙酰化酶活性越强。在NAD+存在的情况下,SIRT 通过脱去下游靶蛋白赖氨酸残基上的乙酰基发挥作用。
SIRT 家族是衰老与寿命的调控因子,在神经退行性疾病、心血管疾病、糖尿病、代谢性疾病等年龄相关性疾病中发挥保护作用[4]。在sir2的7 个同源基因中,目前对SIRT1 的研究最为深入。SIRT1 在组织中分布广泛,在心脏、肾脏、肝脏、大脑、胰腺、骨骼肌的表达较高。SIRT1作用的底物除组蛋白外,还包括过氧化物酶体增殖物激活受体γ共激活因子-1α(PCG-1α)、叉头框转录因子(FoxO)、核因子κB(NF-κB)、p53、内皮型一氧化氮合酶(eNOS)等多种非组蛋白。SIRT1 的表达和活性受多种转录因子、病理应激状态调控。FoxO、E2F 转录因子1、苏素化修饰因子、c-jun 氨基末端激酶(JNK1)等能增加SIRT1 的表达和活性;而p53、过氧化物酶体增殖物激活受体γ(PPARγ)、肿瘤高甲基化基因1 等能抑制SIRT1 的活性和表达[3,5]。
2 SIRT1 在心血管系统中的生理学作用
SIRT1 在心血管系统中发挥着重要作用,调控心肌梗死、心肌肥大、心肌病、血脂异常等心血管疾病的发生与发展。SIRT1 在哺乳动物心脏中高度表达,并通过脱乙酰化作用发挥调节线粒体功能、减轻氧化应激反应、抑制细胞凋亡、抑制炎性反应等作用。
2.1 调节线粒体功能
心脏为血液流动提供动力,需要消耗大量能量,而线粒体是能量的主要来源。线粒体通过氧化磷酸化产生三磷酸腺苷(ATP)为心肌细胞提供能量,并维持细胞内环境稳定。线粒体功能的稳定对于维持心脏正常生理功能至关重要。小鼠心脏缺乏SIRT1 使其表现出与线粒体功能障碍密切相关的进行性扩张型心肌病,心肌细胞缩小,心肌纤维缺失,心肌细胞线粒体肿胀和嵴缺失。心肌细胞增强因子(Mef2)通过乙酰化修饰激活,参与调控心脏分化与线粒体生物发生。小鼠心脏SIRT1 的缺失干扰了Mef2 的乙酰化模式,降低了Mef2a 和Mef2d 的乙酰化水平,同时升高了Mef2c 的乙酰化水平,导致线粒体DNA 含量及其编码基因表达降低,促使线粒体生物发生异常并抑制线粒体呼吸[6]。
PCG-1α是调控线粒体生物发生的核受体转录辅活化子,其水平降低与线粒体生物发生的不足有关,从而导致线粒体功能障碍。SIRT1对PGC-1α发挥脱乙酰基作用使其活化,活化的PGC-1α进一步激活核呼吸因子1/2(NRF-1/2)和线粒体转录因子A(TFAM),增加线粒体的生物发生。在氧化应激状态的大鼠心肌细胞H9c2 中,白藜芦醇通过激活SIRT1 诱导PGC-1α、NRF-1和NRF-2 的表达,而给予SIRT1 抑制剂尼克酰胺后,这些转录因子的表达被抑制[7]。
腺苷酸活化蛋白激酶(AMPK)是调控能量代谢的重要分子。SIRT1 激活LKB1(liver kinase B1),使其下游的AMPK 表达增加。AMPK 进一步促进尼克酰胺磷酸核糖转移酶(Nampt)的转录,使细胞内NAD+水平升高,SIRT1 活性增加。Meng 等[8]对卵巢切除小鼠进行冠状动脉结扎术建立心肌缺血模型,发现白藜芦醇使小鼠缺血心肌的SIRT1 表达和活性增强,AMPK 信号被激活,从而抑制了线粒体通透性转换孔的病理性开放,缓解了线粒体肿胀程度和细胞氧化应激水平,恢复了线粒体功能,减少了缺血性心脏病小鼠心肌细胞的损伤。同时,白藜芦醇通过激活SIRT1/AMPK 信号通路,使线粒体呼吸链酶复合物Ⅰ、Ⅳ、Ⅴ活性恢复,促进了ATP 合成(见图1)。
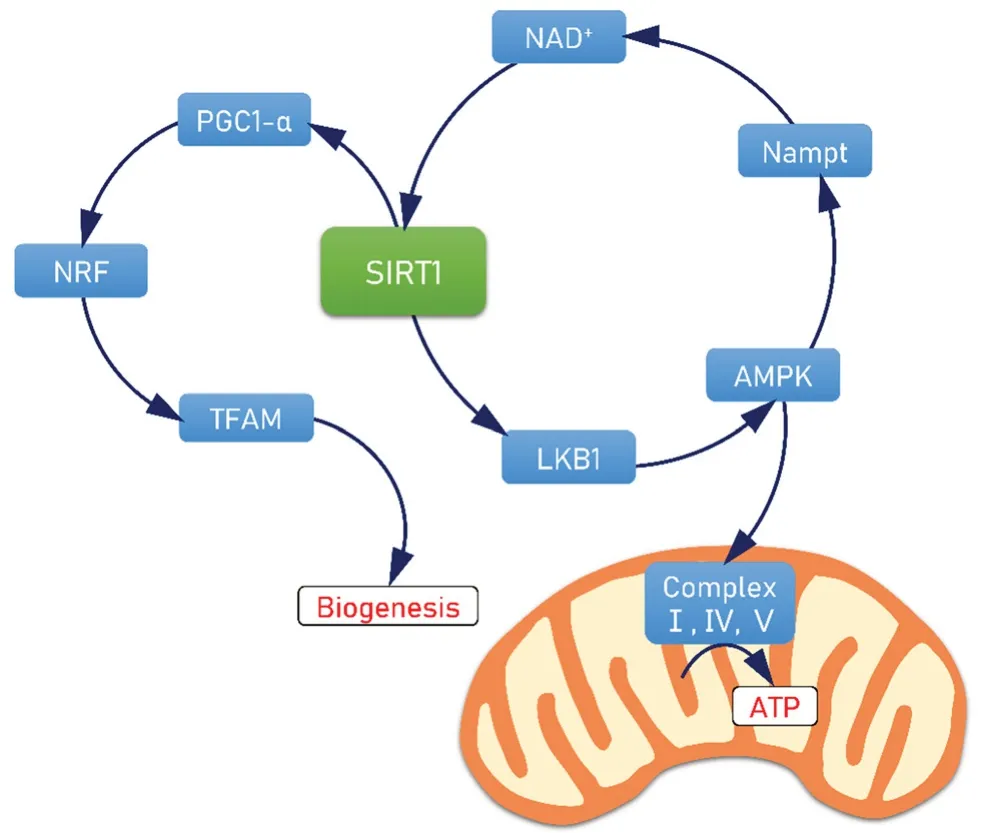
图1 SIRT1 通过PCG-1α 与AMPK 信号通路调节线粒体功能Fig 1 SIRT1 regulated the mitochondrial function through PGC-1α/AMPK pathway
2.2 抑制氧化应激
在病理状态下,线粒体持续产生的活性氧(ROS)蓄积在细胞内,引起氧化应激,破坏细胞稳态,诱导细胞发生死亡级联反应。ROS 过量生成是诱导细胞死亡的重要生理机制。SIRT1 对细胞内氧化还原平衡状态具有高度敏感性,可通过多种途径减少ROS 生成,从而保护心血管功能。
超氧化物歧化酶(SOD)是一类具有抗氧化作用的金属酶,在抵御ROS 损伤中发挥重要作用。有研究表明,白藜芦醇增加细胞核中SIRT1的表达,诱导锰超氧化物歧化酶(Mn-SOD)的生成,缓解心力衰竭小鼠心肌的氧化应激损伤;而在心肌特异性SIRT1 过表达小鼠中,SIRT1 通过刺激FoxO1 的转录活性,上调Mn-SOD 的表达,抑制氧化应激,促进心肌细胞存活[9]。Olmos等[10]报道,SIRT1 参与调节血管内皮细胞中FoxO3a/PGC-1α复合物的形成,使Mn-SOD、过氧化氢酶、过氧化物酶5 和硫氧还蛋白还原酶2等氧化应激保护因子的mRNA 和蛋白水平升高。
2.3 抑制炎症反应
炎症反应是细胞损伤或应激使免疫系统受到激活后机体产生的防御反应。SIRT1 能够抑制NF-κB 炎症信号通路,减少炎性因子释放,从而减轻炎症反应。NF-κB 受到SIRT1 调控,SIRT1与NF-κB 的P65/RelA 亚单位结合,降低P65/RelA 乙酰化程度,使NF-κB 失活,减少其下游肿瘤坏死因子-α(TNF-α)、白细胞介素-1β(IL-1β)等炎性因子的表达[11]。Li 等[12]发现,对于心脏缺血/再灌注损伤小鼠,应用咖啡酸邻硝基苯乙酯(CAPE-o NO2)后SIRT1 表达增加,并通过NF-κB 途径抑制了心脏组织中ROS 生成和炎性因子的表达,减少缺氧复氧诱导的氧化应激与炎症反应。
2.4 抑制细胞凋亡
细胞凋亡是一种自主有序的程序性死亡,过高的炎性细胞因子或ROS 水平可诱导细胞进入凋亡。心肌细胞凋亡是心力衰竭等多种心脏疾病发生发展的重要病理基础。心肌细胞的不可再生性使其在发生凋亡时数量减少,从而损害心脏功能。心脏特异性SIRT1 过表达使氧化应激下心肌细胞的凋亡显著降低,心肌损伤减轻。Yuan 等[13]的研究发现,阿霉素(DOX)降低了补体C1q/肿瘤坏死因子相关蛋白3(CTRP3)的表达,而心脏特异性过表达CTRP3 激活了SIRT1,从而抑制炎症反应和心肌细胞凋亡,减轻了DOX 诱导的心功能不全。
SIRT1 可通过FoxO1 信号通路,使抗凋亡蛋白Bcl-xL 表达增加,同时下调促凋亡蛋白caspase-3 和Bcl 相关X 蛋白(Bax),抑制心肌细胞凋亡。而Chen 等[14]报道,在缺氧H9c2 细胞中给予白藜芦醇,SIRT1 对FoxO1 的功能具有双重影响,SIRT1 经FoxO1 信号通路,促进了超氧化物歧化酶Mn-SOD 的转录,增加了Mn-SOD 表达,抑制了细胞凋亡;同时,细胞周期依赖性激酶特异性抑制蛋白p27KIP1被激活,使FoxO1 诱导细胞周期停滞的作用增强。
p53 在调控细胞周期与凋亡中发挥重要作用,其活性受Bcl 相关X 蛋白(Bax)介导。SIRT1 可降低p53 的乙酰化程度,抑制Bax 启动子与p53的结合,下调Bax 的表达,减少p53 依赖途径的细胞凋亡。
此外,Luo 等[15]报道,在低氧诱导的H9c2细胞中,肌醇依赖性激酶1α(IRE1α)被激活,而SIRT1 过表达可降低IRE1α活性,抑制IRE1α通过JNK/c-jun 和NF-κB/p65 途径介导的H9c2 心肌细胞凋亡。
2.5 调控细胞自噬
自噬是一种依赖于溶酶体的常见细胞代谢过程,对于维持细胞正常功能有着重要的作用。根据底物的选择性,可以将自噬分为选择性自噬和非选择性自噬,前者包括线粒体自噬、内质网自噬、核糖体自噬等[16]。在人类心肌细胞缺氧/复氧损伤后,提高SIRT1 表达可以激活线粒体自噬,减少心肌细胞损伤[17-18]。此外,Silva 等[19]发现,衣霉素或异丙肾上腺素诱导的内质网应激反应中,SIRT1 缺乏小鼠表现出自噬抑制和心功能不全加重,进一步研究发现,SIRT1 可激活eEF2K/eEF2 通路增强心肌细胞的内质网自噬,减轻衣霉素诱导的内质网应激反应,从而促进心肌细胞存活。
3 SIRT1 在CVD 中的作用
CVD 的发展是一个连续的过程,涉及氧化应激、炎症、细胞凋亡与自噬等多种机制。心肌细胞不可再生,因此延长心肌细胞的寿命、维持心肌细胞活力,对心脏疾病的防治至关重要。SIRT1 通过参与调节细胞的能量代谢,减少氧化应激条件下细胞的凋亡和衰老,促进细胞自噬,最终保护心血管功能,减轻CVD 的发展。研究表明,SIRT1 可调控心力衰竭、心肌缺血再灌注损伤、心肌肥厚、糖尿病心肌病、动脉粥样硬化等CVD 的发生与过程。
3.1 对心力衰竭及左心室功能的影响
心力衰竭是常见的心血管疾病终末期临床表现,展现出复杂的表型,如心肌细胞死亡、心肌纤维化、心肌收缩力降低等。心力衰竭的发生涉及到氧化应激、心肌细胞凋亡、肌浆网Ca2+-ATPase(SERCA2a)功能不足等机制。在晚期心力衰竭患者的心肌组织中,SIRT1 表达显著减少,Mn-SOD 及抗凋亡分子Bcl-xL 受到明显抑制,而促凋亡分子Bax 的表达明显增加;SIRT1 的缺乏可能通过p53 和FoxO1 信号通路,介导了心肌细胞凋亡[20]。SIRT1 激动剂白藜芦醇能够增加心肌细胞核SIRT1 表达,促进Mn-SOD 生成,保护心肌免受氧化应激损伤。
SERCA2a 是调节细胞内游离Ca2+浓度的酶,其表达或活性不足直接影响细胞内Ca2+稳态,降低心肌收缩力,从而引发心力衰竭。Gorski 等[21]的研究表明,在多种动物的衰竭心脏中,SIRT1激动剂增强了SERCA2a 的活性,进一步研究发现,SIRT1 可能通过对SERCA2a 赖氨酸残基K492 的去乙酰化作用,促进了ATP 与SERCA2a的结合,从而恢复衰竭心脏的心肌功能。
临床试验也证实了SIRT1 激动剂能够改善心功能。Magyar 等[22]发现,SIRT1 激动剂白藜芦醇(10 mg·d-1,持续3 个月)能够显著改善稳定型冠心病患者的左心室舒张功能,左心室收缩功能也略有改善。Militaru 等[23]的研究也表明,口服白藜芦醇(20 mg·d-1,持续60 d)能够显著降低稳定型心绞痛患者的B 型利钠肽水平,改善左心室功能,减少心绞痛发生次数;而白藜芦醇与果糖硼酸钙联用时,对心绞痛患者的生活质量改善更为明显。
3.2 对心肌缺血再灌注损伤的影响
冠状动脉阻塞是诱发缺血性心脏病的主要原因。恢复冠状动脉的血流供给可减轻心肌的缺血缺氧状态以及心肌坏死,但心肌缺血再灌注(I/R)常常会加剧心肌损伤。SIRT1 通过调节I/R 过程中的主要病理过程,如氧化应激、炎症、细胞凋亡等,减轻I/R 损伤,改善心肌功能。在心脏I/R 小鼠模型中,心脏特异性SIRT1 敲除使心肌梗死面积和心肌细胞凋亡率显著增加,而心脏特异性SIRT1 过表达减少了心肌梗死面积和心肌细胞凋亡;进一步研究发现,SIRT1 上调了Mn-SOD、硫氧还蛋白1、Bcl-xL 等促存活分子的表达,下调了促凋亡分子Bax 和活化的caspase-3,同时刺激了FoxO1 的转录活性,抑制了心肌细胞的氧化应激,保护心脏免受I/R 损伤[24]。应用SIRT1 激动剂也能产生类似生物学的效应,白藜芦醇可通过激活SIRT1,减少心肌梗死面积,降低I/R 导致的心肌损伤;而SIRT1 抑制剂尼克酰胺以剂量依赖性方式,加重心肌细胞凋亡。缺血后处理(IPC)是减轻I/R 损伤的有效方式之一,SIRT1在IPC 中的有益作用与PI3K/Akt 信号通路有关,SIRT1 的去乙酰化作用使Akt 活化,促进Akt 与PIP3 结合,SIRT1 通过激活PI3K/Akt 途径,恢复了IPC 介导的心脏保护作用[25]。
3.3 对心肌肥厚的影响
心脏长期负荷过度导致心肌细胞体积增大,心肌厚度和重量增加。在心脏压力超负荷和氧化应激条件下,SIRT1 水平显著上调。SIRT1 在抑制心肌肥厚中的作用较为复杂。自发性高血压大鼠的心脏SIRT1 表达升高,心肌细胞内SIRT1 mRNA 与蛋白表达及心肌肥厚程度成正相关。此外,抑制SIRT1 表达可以减少压力超负荷所引起的心肌肥厚和心力衰竭;而PPARγ和SIRT1 表达同时增加,会加重心功能不全[26]。但是,Li 等[27]报道SIRT1 减少了蛋白激酶C-ζ(PKC-ζ)的乙酰化,降低了PKC-ζ途径诱导的心肌肥厚。
3.4 对糖尿病心肌病的影响
糖尿病心肌病是指在心肌代谢紊乱及心脏微血管病变的基础上,导致的心肌广泛灶性坏死,与线粒体的生物发生功能受损有关。Ma 等[28]研究表明,心脏特异性SIRT1基因敲除小鼠表现出糖尿病心肌病的症状,包括心肌肥厚和功能障碍、胰岛素抵抗和糖代谢异常;进一步研究发现,白藜芦醇激活了SIRT1,SIRT1 通过介导PGC-1α的去乙酰化,诱导NRF-1/2、ERR-α和TFAM 的表达增加,逆转了糖尿病心肌病小鼠的线粒体生物发生和线粒体功能,减轻心脏舒张功能障碍。
糖尿病导致心肌eNOS 表达降低,NO 合成受损,诱导氧化应激和细胞凋亡。Fourny 等[29]研究表明,在糖尿病大鼠左心室心肌注射腺病毒载体Ad-SIRT1 能上调心脏SIRT1 表达,减少心肌细胞内eNOS 乙酰化,增加eNOS 磷酸化;SIRT1过表达改善了糖尿病大鼠的心功能,缩小了心肌梗死面积,减轻了心肌缺血再灌注损伤和氧化应激。Koka 等[30]研究表明,在糖尿病小鼠缺血再灌注的心脏中,SIRT1 的过表达不仅抑制心肌细胞超氧化物的产生,还提高了抗氧化酶SOD 的活性。而SIRT1 在糖尿病心肌病中的抗凋亡作用与FoxO 有关。在自发性糖尿病Goto-Kakizaki 大鼠中,SIRT1 激活FoxO3a 信号通路的同时脱去了p53 赖氨酸373/382 位点的乙酰基,抑制了p53 活性,从而减轻了糖尿病心脏的重塑和心肌细胞凋亡[31]。此外,糖尿病心肌功能受损与SERCA2a的水平降低有关。Sulaiman 等[32]报道,糖尿病小鼠心脏的SIRT1 活性降低,使SERCA2a 下调;白藜芦醇能激活SIRT1 增强SERCA2a 表达,从而改善糖尿病小鼠的心脏功能。
临床研究表明,糖尿病冠心病患者补充白藜芦醇(500 mg·d-1,持续4 周)可使外周血单个核细胞的PPAR-γ和SIRT1 上调,患者对胰岛素的敏感性显著升高,空腹血糖、胰岛素、胰岛素抵抗显著降低[33]。
3.5 对动脉粥样硬化的影响
动脉粥样硬化的发生与泡沫细胞形成密切相关。泡沫细胞主要来源于由单核细胞分化成的巨噬细胞以及血管平滑肌细胞(VSMC)。这两种来源的泡沫细胞的形成都可被SIRT1 抑制。SIRT1通过介导肝X 受体(LXR)赖氨酸K432 的脱乙酰基,促进胆固醇逆向转运,清除巨噬细胞中的胆固醇,抑制巨噬源性泡沫细胞形成。此外,SIRT1 还通过抑制NF-κB 信号通路,减少氧化型低密度脂蛋白(oxLDL)受体-1 的表达,降低巨噬细胞对oxLDL 的摄取,从而减少巨噬源性泡沫细胞的形成[34]。而在VSMC 中,SIRT1 功能的降低使NF-κB 活性增强,促进oxLDL 诱导细胞内胆固醇和脂质滴的积累,增加了VSMC 源性泡沫细胞的形成和迁移能力;在给予SIRT1 激活剂STR1720 后,SIRT1 的表达上调,NF-κB 信号通路受到抑制,SRT1720 剂量依赖性地减少了oxLDL 诱导的VSMC 源性泡沫细胞的形成[35]。
此外,血脂异常也是动脉粥样硬化的诱发因素之一。而SIRT1 信号通路的激活能够改善血脂水平。在ApoE-/-小鼠中,SIRT1 激动剂SRT3025 通过抑制前蛋白转化酶枯草溶菌素9(Pcsk9)的表达,使血浆总胆固醇、低密度脂蛋白胆固醇和极低密度脂蛋白胆固醇显著降低,主动脉斑块明显缩小,抑制动脉粥样硬化的发生[36]。临床研究显示,健康吸烟人群口服SIRT1激动剂SRT2104(2.0 g·d-1,持续28 d)能够降低血清总胆固醇、低密度脂蛋白胆固醇和三酰甘油浓度[37]。而糖尿病冠心病患者补充白藜芦醇(500 mg·d-1,持续4 周)后,高密度脂蛋白胆固醇水平显著增加,总胆固醇与高密度脂蛋白胆固醇的比值显著降低[33]。
3.6 对其他CVD 的影响
Zhou 等[38]发现,在血管紧张素Ⅱ(AngⅡ)诱导的高血压小鼠中,SIRT1 表达和脱乙酰基活性显著降低,下游靶蛋白p53 的乙酰化水平显著升高;进一步研究发现,在VSMC 中,SIRT1 过表达能够抑制AngⅡ输注引起的收缩压升高,降低TGF-β1 启动子特异性结合位点上NF-κB 的结合,从而减轻胸主动脉和肾主动脉的血管重塑。而AngⅡ参与诱导基质金属蛋白酶(MMP)活性升高,刺激主动脉炎症反应,诱发主动脉夹层。SIRT1 通过介导NF-κB 和内源性MMP 拮抗剂TIMP-1 的去乙酰化,抑制MMP 基因活性和转录,在受到氧化应激和炎性刺激时维持主动脉壁结构的完整性,从而减少主动脉夹层的发生[39]。
衰老使血管内皮SIRT1 功能部分丧失,NOsGC-cGMP 信号传导受损,血管舒张功能下降。增强老年小鼠血管内皮SIRT1 的表达后,COX-2介导的血管收缩反应受到抑制,血管平滑肌细胞中sGCβ1 的表达和活性增加,缓解了年龄诱导的血管舒张反应减弱[40]。
4 展望
现有研究证实,在心力衰竭、心肌缺血再灌注损伤、心肌肥厚、糖尿病心肌病、动脉粥样硬化等疾病的病理过程中,SIRT1 能够减少细胞损伤,改善心血管功能。SIRT1 作为能够改善心血管功能的靶点,其激动剂作为潜在的治疗药,有望应用于多种CVD,具有一定的开发价值。目前,临床前研究已经展现了SIRT1 激动剂对CVD的治疗作用,但临床研究的数量以及样本量仍然有限。但是,SIRT1 表达过高(12.5 倍)时会导致心肌细胞凋亡,心功能减退[41]。而且,SIRT1可以减少转录因子核因子E2 相关因子2(Nrf2)乙酰化以及依赖Nrf2 的相关抗氧化基因转录[42],可能会加重心脏损害,这些使得SIRT1 激动剂的应用受到一定的局限。综上所述,研究SIRT1 在心血管中的作用机制,开发以SIRT1 为靶点的药物,将为心脏疾病的预防与治疗提供新的思路与方案。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
世界科学技术-中医药现代化(2022年2期)2022-05-25
云南化工(2021年11期)2022-01-12
世界科学技术-中医药现代化(2021年7期)2021-11-04
海南医学(2016年8期)2016-06-08
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国病理生理杂志(2015年8期)2015-12-21
中国当代医药(2015年16期)2015-03-01
中国药理学通报(2014年2期)2014-05-09
现代检验医学杂志(2014年6期)2014-02-02
