
齐墩果酸A环并咪唑类衍生物的合成及活性研究
2021-02-11 03:13张蓬勃宋艳玲
沈阳化工大学学报 2021年4期
张蓬勃,宋艳玲
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
齐墩果酸(oleanolic acid,OA)是分布于自然界中的齐墩果烷型五环三萜类天然产物[1],不仅具有良好的肝脏保护生物活性[2]、抗炎[3]、抗病毒[4-5]、降血糖[6]、降血脂[7]等,而且具有很强的抗肿瘤作用[8].由于其分子结构刚性强、水溶性差等原因,因此,通过对其进行适当的化学结构修饰,以期得到具有较强抗肿瘤活性的衍生物是现今主要的研究目的.肺癌、肝癌为常见恶性肿瘤,具有较高发病率和死亡率.本文以人肺癌细胞(A549)和人肝癌细胞(HepG2)为生物模型,测试化合物的生物学活性.
咪唑环具有特殊的化学结构,其能够容易地与生物体内蛋白受体和酶等形成氢键,因此,咪唑结构作为一种优良的化学骨架在药学领域得到了广泛的研究[9].研究发现,母核引入咪唑的化合物,其生物活性大幅提升,具有广泛的生物活性,如作为质子泵抑制剂、组胺受体拮抗剂、抗寄生虫、抗肿瘤等,在临床应用上具有良好的应用前景.已上市的抗肿瘤药物,如维利帕尼、达卡巴嗪、硫唑嘌呤等均具有咪唑活性片段(图1).本文以齐墩果酸为原料,通过化学合成在A环引入咪唑结构,28位结构修饰合成Ⅰ1、Ⅱ1-2两类齐墩果酸衍生物.

图1 咪唑类已上市抗肿瘤药物Fig.1 Imidazoles have been marketed as anti-tumor drugs
1 实验部分
1.1 仪器与试剂
Bruker ARX-600型核磁共振仪,美国Bruker公司;Büchi B-540熔点测定仪,瑞士Büchi公司;薄层色谱(GF-254)、柱色谱硅胶(200~300目),青岛海洋化工有限公司;齐墩果酸(质量分数98%),陕西慈缘生物技术有限公司;芳香胺类药品,上海麦克林化学试剂有限公司.其他溶剂和药品均为分析纯.
1.2 合成实验
OA衍生物的合成路线见图2.

试剂和反应条件:(a) 琼斯试剂,丙酮,异丙醇,0 ℃; (b) 二氧化硒,乙酸,118 ℃; (c) 甲酰胺,100 ℃; (d) 碳酸钾,碘甲烷,N,N-二甲基甲酰胺,室温; (e) 草酰氯,芳香胺,室温.图2 OA衍生物的合成路线Fig.2 Synthetic route of OA derivatives
1.2.1 3-羰基-齐墩果酸(1)的制备
将齐墩果酸 OA(4.40 g,9.35 mmol)溶于100 mL丙酮中,冰浴下缓慢滴加Jones试剂11.5 mL,室温下反应2 h,TLC监测反应.反应完毕,加入130 mL异丙醇淬灭.反应结束后,减压蒸除部分溶剂,乙酸乙酯萃取,饱和NaCl溶液洗涤,水洗,合并有机相,无水硫酸钠干燥,抽滤,减压蒸干,甲醇重结晶得白色晶体化合物(1)4.15 g,产率95%,mp 186~187 ℃(文献mp 185~186 ℃)[10].
1.2.2 2,3-二羰基-齐墩果酸(2)的制备
将3-羰基-齐墩果酸(4.15 g,8.87 mmol)溶于50 mL无水乙酸中,加入二氧化硒(0.118 g,1.07 mmol)回流24 h,TLC监测反应.反应结束后,旋蒸除去乙酸溶剂,二氯甲烷萃取,饱和Na2CO3溶液洗涤,合并有机相,无水硫酸钠干燥,抽滤,减压蒸干,得白色固体化合物(2) 3.16 g,产率76.3%,mp 191~193 ℃.
1.2.3 A环并咪唑齐墩果酸(3)的制备
将2,3-二羰基-齐墩果酸(1.00g,2.14 mmol)溶于20 mL甲酰胺中,100 ℃下反应4 h,TLC监测反应.反应结束后,加入适量二氯甲烷稀释,饱和NaCl溶液洗涤,合并有机相,无水硫酸钠干燥,抽滤,减压蒸干,得白色固体化合物(3) 0.66 g,产率66%,mp 232~235 ℃.
1.2.4 化合物Ⅰ1的制备
将A环并咪唑齐墩果酸(0.35 g,0.732 mmol)和无水碳酸钾(0.253 g,1.83 mmol)溶于20 mL干燥的N,N-二甲基甲酰胺中,搅拌下缓慢加入碘甲烷(0.208 g,1.46 mmol),室温下继续搅拌反应2 h,TLC监测反应.反应结束后加水稀释,乙酸乙酯萃取,饱和NaCl溶液洗涤合并有机相,无水硫酸钠干燥,抽滤,减压蒸干溶剂,硅胶柱层析分离(200~300目,洗脱剂:石油醚/乙酸乙酯),得到白色固体产物化合物Ⅰ10.162 g,产率46.29%,mp 240~244 ℃.1H-NMR(600 MHz,DMSO),δ:12.50 (s,1H),8.01 (s,1H),5.29 (s,1H),3.63 (s,3H),1.15 (s,3H),1.01 (s,3H),0.98 (s,3H),0.94 (s,3H),0.91 (s,3H),0.82 (s,3H),0.77 (s,3H).
1.2.5 化合物Ⅱ1~2的制备
1.2.5.1 化合物Ⅱ1的制备
将A环并咪唑齐墩果酸(0.30 g,0.627 mmol)溶于20 mL干燥二氯甲烷溶液中,冰浴条件下缓慢滴加草酰氯(0.399 g,3.14 mmol),0.5 h后撤冰,40 ℃下反应2~3 h,TLC监测反应.反应结束后蒸发有机溶剂,干燥环己烷冲洗3次.将其溶于20 mL无水二氯甲烷中,缓慢滴入溶于10 mL苯胺(0.293 g,3.14 mmol)的二氯甲烷溶液,加入干燥吡啶(0.5 mL)、DMAP(0.001 4 g,0.04 mmol)在25~30 ℃条件下反应12 h,TLC监测反应.反应结束后二氯甲烷萃取,蒸馏水洗涤,合并有机相,无水硫酸钠干燥,抽滤,减压蒸干,硅胶柱色谱分离,甲醇/二氯甲烷洗脱,得白色固体化合物 Ⅱ10.131 g,产率43.6%,mp 211~212 ℃.1H-NMR (600 MHz,DMSO),δ:12.52 (s,1H),9.29 (s,1H),8.00 (s,1H),7.74~7.72 (m,2H),7.41~7.37 (m,3H),5.31 (s,1H),1.16 (s,3H),1.01 (s,3H),0.98 (s,3H),0.93 (s,3H),0.90 (s,3H),0.78 (s,3H),0.76 (s,3H).
1.2.5.2 化合物Ⅱ2的制备
实验步骤同化合物Ⅱ1的制备,A环并咪唑齐墩果酸(0.30 g,0.627 mmol)与对氟苯胺(0.349 g,3.14 mmol)反应得到淡黄色固体化合物Ⅱ20.118 g,产率39.3%,mp 215~218 ℃.1H-NMR (600 MHz,DMSO),δ:12.52 (s,1H),9.52 (s,1H),8.00 (s,1H),7.76~7.73 (m,2H),7.39~7.37 (m,2H),5.34 (s,1H),1.14 (s,3H),1.01 (s,3H),0.98 (s,3H),0.94 (s,3H),0.90 (s,3H),0.79 (s,3H),0.76 (s,3H).
2 抗肿瘤活性实验
以维利帕尼为阳性对照药,维利帕尼结构中含有咪唑活性片段,其抑制肿瘤细胞活性具有参考意义.取对数生长期的人肺癌细胞(A549)和人肝癌细胞(HepG2),以每孔5×103个接种于96孔板内,每组3个复孔,CO2培养箱中培养24 h后备用.待细胞贴壁后,分别加入待测化合物、OA和维利帕尼,药品浓度为10 μmol/L,空白组为阴性对照组,维利帕尼为阳性对照组.培养箱中继续培养72 h.加入MTT溶液(5 g/mL,20 μL),继续培养4 h,吸去每孔上清液,加入二甲基亚砜200 μL,震荡器上震荡5 min.然后用酶标仪于570 nm处测OD值,重复3次实验,取平均值,计算各组细胞的IC50.抑制率=[(对照组OD值-药物组OD值)/对照组OD值]×100%.化合物抗肿瘤活性和抑制率见表1.
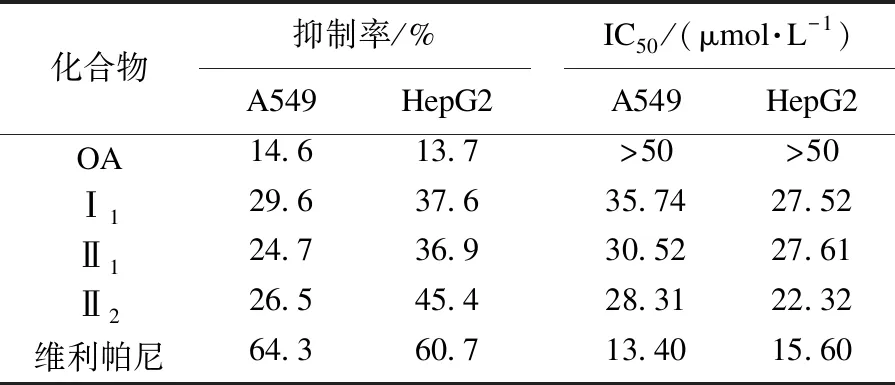
表1 化合物对A549和HepG2细胞株的抗肿瘤活性Table 1 Anti-tumor activity of the target compounds on A549 and HepG2 cell lines
表1中数据显示,化合物Ⅱ2对A549、HepG2细胞具有良好的生长抑制作用,取代基苯环上取代官能团的改变,使其抑制率具有差别.引入氟原子,其活性增强(22.32 μmol/L),无取代基苯环活性较低(27.61 μmol/L).设计合成的3个化合物均优于原料OA.
3 结 论
设计并合成了3个未见文献报道的齐墩果酸衍生物.活性试验表明,设计合成的化合物对A549和HepG2肿瘤细胞均有明显的抑制生长作用.这表明引入咪唑活性片段提高了其抗肿瘤活性,与设计思路相符,尤其是化合物Ⅱ2对A549、HepG2细胞具有良好的抑制作用.以维利帕尼为阳性对照药物,进一步说明了咪唑活性基团的抗肿瘤活性优点.由于实验条件有限,对于咪唑活性基团的研究不足,对于该类化合物构效关系正在进一步研究.本研究结果对齐墩果酸衍生物的进一步优化设计具有一定的指导意义.
猜你喜欢
小作家报·教研博览(2022年11期)2022-04-02
草业科学(2022年2期)2022-03-21
皮肤病与性病(2021年3期)2021-07-30
健康博览(2021年4期)2021-04-23
爱你(2019年25期)2019-07-16
科技视界(2016年26期)2016-12-17
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
中国质量万里行(2015年1期)2015-01-27
湖北农业科学(2011年16期)2011-11-18
