
积雪草酸新型类似物的合成及体外抗肿瘤活性研究
2021-02-11 03:13:20林碧琦孟艳秋
沈阳化工大学学报 2021年4期
林碧琦,孟艳秋
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
积雪草酸(asiatic acid,AA)又名亚细亚酸,存在于丁龙脑香科植物龙脑香的树脂和挥发油巾中,也可由伞形科植物积雪草中的积雪草苷水解而得,是一种五环三萜类化合物,具有乌苏烷型结构,不溶于水,易溶于DMF、THF等有机溶剂[1-3].积雪草酸具有广泛的药理活性,如抗肿瘤、神经保护、抗氧化、抗抑郁、保护肝脏,以及抗炎等作用[4-7].有研究发现,对积雪草酸进行结构修饰可以提高其抗肿瘤活性.Gonçalves[8]等通过对积雪草酸的A环进行改造,得到了化合物1、2,并且选用HT-29和HeLa细胞系评估其抗肿瘤活性,结果显示目标化合物的活性优于母体积雪草酸.Huang等[9]将积雪草酸C-2、C-3、C-23位羟基改造为乙酰氧基,在C-28位引入不同的氨基膦酸酯,并采用MTT法检测化合物对癌细胞的抑制活性,结果表明该化合物与母体积雪草酸相比抗肿瘤活性明显提高.基于课题组前期工作积累[10-12],笔者以积雪草酸为先导化合物进行一系列结构改造,提高积雪草酸抗肿瘤活性.
由于积雪草酸水溶性不好、生物利用度低等因素,限制了其在抗肿瘤方面的应用.笔者在保留积雪草酸五环三萜骨架的基础上,以积雪草酸为先导化合物对其A环上的3个羟基、C-12位不饱和双键,以及C-28位的羧基进行结构修饰与改造,设计合成了8个积雪草酸衍生物,以期得到活性更高的化合物.
1 仪器与试剂
Büchi B-540熔点测定仪,Büchi公司;Bruker ARX-300型核磁共振分析仪,CDCl3为溶剂,TMS为内标,Bruker公司;积雪草酸,纯度95%(质量分数),南京狄尔格医药科技有限公司;柱层析色谱用200~300目硅胶,薄层色谱用GF254,青岛海洋化工厂,显色剂为体积分数分数10%的硫酸乙醇溶液,实验室自制;活性测试所用的RPMI-1640培养基(体积分数10%的胎牛血清,100 U/mL青霉素,100 mg/L链霉素)、四甲基偶氮唑盐(MTT)、蛋白酶 K(proteinase K)和小牛血清蛋白(BSA),上海图赫实业有限公司;活性测试用人癌细胞HepG2和A549,沈阳药科大学药理实验室;阳性对照物吉非替尼(Gefitinib)和阿霉素(Adriamycin),阿斯利康制药有限公司.所有试剂均为分析纯或化学纯.
2 合成路线
目标化合物的合成路线如图1所示.
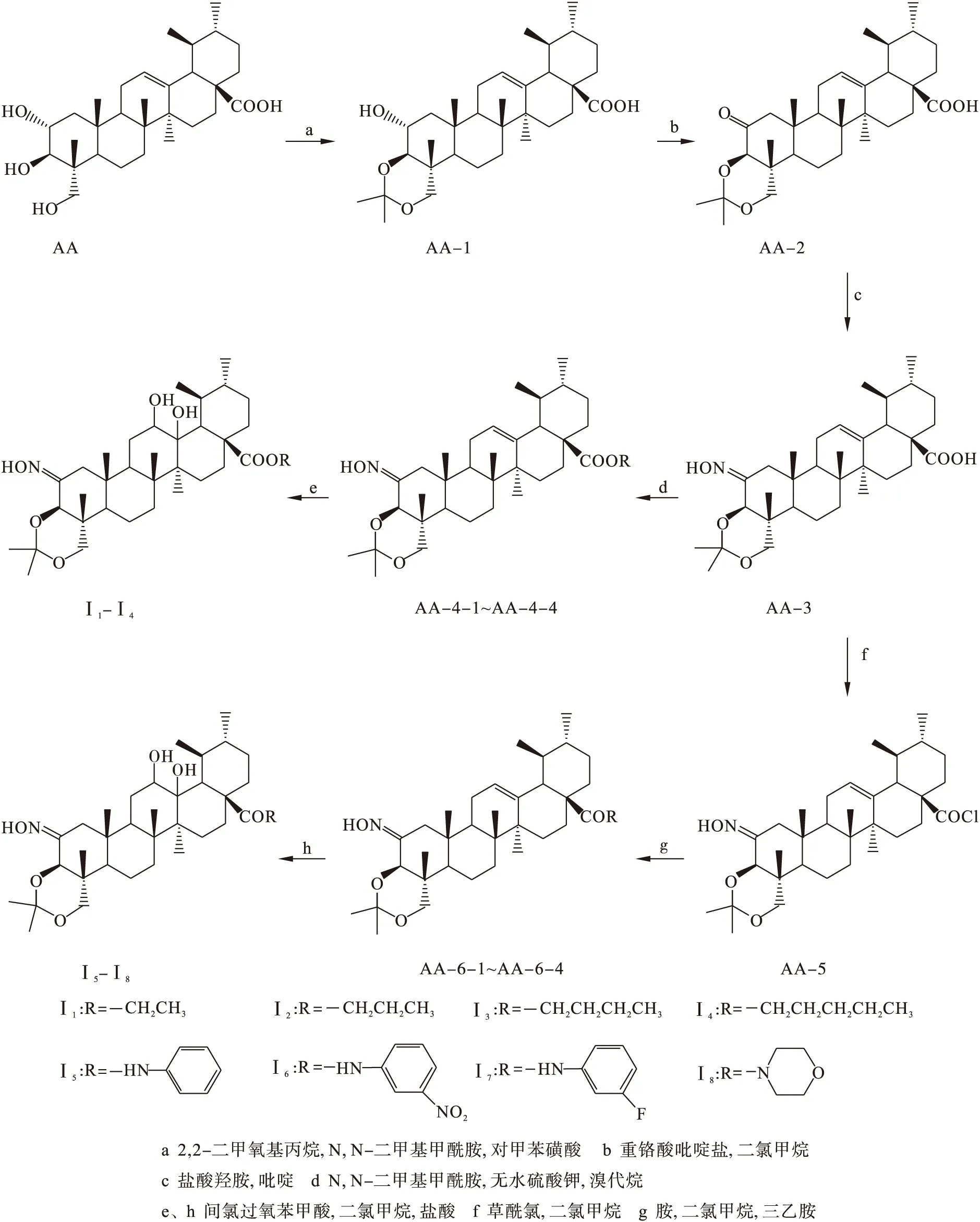
图1 目标化合物的合成路线Fig.1 Synthetic route of target compound
3 目标化合物的合成
3.1 2-羟基-3β,23-O-异亚丙基-12-烯-28-羧酸的合成(AA-1)
取AA(100 mg 0.19 mmol)充分溶解在20 mL 的DMF中,依次加入对甲基苯磺酸(17.0 mg,0.10 mmol)和2,2-二甲氧基丙烷(0.5 mL,2.0 mmol),室温下反应,TLC检测反应终点[V(石油醚)∶V(乙酸乙酯)=3∶1].用质量分数5%的NaOH调节pH为7~8,用乙酸乙酯和饱和食盐水萃取3次,合并有机相,无水硫酸钠干燥过夜,抽滤,减压蒸馏得白色固体AA-1,产率为64.2%.
3.2 2-羰基-3β,23-O-异亚丙基-12-烯-28-羧酸的合成(AA-2)
取AA-1(100 mg,0.19 mmol)置于50 mL茄形瓶中,溶于10 mL二氯甲烷中,加入负载硅胶的重铬酸吡啶鎓盐(450 mg),室温反应2 h,TLC检测反应终点[V(石油醚)∶V(乙酸乙酯)=3∶1].反应结束后,抽滤除去硅胶,乙酸乙酯和饱和食盐水萃取3次,合并有机相,无水硫酸钠干燥过夜,抽滤,减压蒸馏得白色固体AA-2,产率为72.5%.
3.3 2-肟基-3β,23-O-异亚丙基-12-烯-28-羧酸的合成(AA-3)
将中间产物AA-2(100 mg,0.18 mmol)在吡啶中完全溶解,加入15 mg盐酸羟胺,在115 ℃下回流1.5 h,TLC检测反应终点.反应结束后,加入25 mL水搅拌10 min,析出白色固体,减压抽滤,得到化合物AA-3,产率73.2%.
3.4 2-肟基-3β,23-O-异亚丙基-12,13-二羟基-28-乙酯的合成(Ⅰ1)
将中间体AA-3(100 mg,0.18 mmol)溶解在DMF(20 mL)中,依次加入K2CO3(70 mg,0.5 mmol)和溴乙烷(0.2 mL),室温反应6 h,TLC检测反应终点.乙酸乙酯和饱和食盐水萃取3次,合并有机相,无水硫酸钠干燥过夜,抽滤,减压蒸除溶剂,得白色固体AA-4-1.使用CH2Cl2(10 mL)溶解中间产物AA-4-1,加入间氯过氧苯甲酸(75 mg 0.44 mmol),在室温条件下反应8 h,TLC监测反应终点.用二氯甲烷和饱和食盐水反复萃取3次,然后用饱和NaHCO3洗涤至中性,无水硫酸钠干燥,减压蒸除得到白色固体.白色固体用10 mL氯仿溶解,2 mL浓盐酸搅拌2 h,TLC检测反应终点.用饱和食盐水和CHCl3萃取3次,加入少量NaHCO3溶液洗至中性,减压,蒸除溶剂,粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=10∶1,得到化合物Ⅰ1,产率为30.5%.mp 224.2~228.8 ℃.ESI-MS(m/z):604.1(M+H)+.1H-NMR(300 MHz,CDCl3),δ:11.40(s,1H,NOH),4.07(t,J=7.9 Hz,2H,COOCH2CH3),3.88(s,1H,H-3),3.60(d,2H,J=9.6 Hz,H-23),3.49(s,1H,H-12),1.19~1.11(m,3H,COOCH2CH3),1.09(s,3H,CH3),1.07(s,3H,CH3),0.91(d,J=8.8 Hz,3H,CH3),0.88(s,3H,CH3),0.85(s,3H,CH3),0.78(s,3H,CH3).
3.5 2-肟基-3β,23-O-异亚丙基-12,13-二羟基-28-丙酯的合成(Ⅰ2)
按照制备Ⅰ1的方法,将中间体AA-3(100 mg,0.18 mmol)与溴丙烷(0.2 mL)反应,粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=10∶1,得到化合物Ⅰ2,产率为27.6%.mp 227.4~232.6 ℃.ESI-MS(m/z):617.7(M+H)+.1H-NMR(300 MHz,CDCl3),δ:11.39(s,1H,NOH),4.42(t,J=7.9 Hz,2H,COOCH2CH2CH3),3.85(s,1H,H-3),3.59(d,2H,J=9.6 Hz,H-23),3.47(s,1H,H-12),1.71~1.55(m,2H,COOCH2CH2CH3),1.25(t,J=7.1 Hz,3H,COOCH2CH2CH3),1.23(s,3H,CH3),1.07(s,3H,CH3),0.94(d,J=8.8 Hz,3H,CH3),0.87(s,3H,CH3),0.85(s,3H,CH3),0.81(s,3H,CH3).
3.6 2-肟基-3β,23-O-异亚丙基-12,13-二羟基-28-丁酯的合成(Ⅰ3)
按照制备Ⅰ1的方法,将中间体AA-3(100 mg,0.18 mmol)与溴丁烷(0.2 mL)反应,粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=10∶1,得到化合物Ⅰ3,产率为28.6%.mp 231.3~235.9 ℃.ESI-MS(m/z):631.8(M+H)+.1H-NMR(300 MHz,CDCl3),δ:11.43(s,1H,NOH),4.22(t,J=7.9 Hz,2H,COOCH2CH2CH2CH3),3.91(s,1H,H-3),3.59(d,2H,J=9.6 Hz,H-23),3.47(s,1H,H-12),1.84(t,J=7.1 Hz,3H,COOCH2CH2CH2CH3),1.77~1.34(m,4H,COOCH2(CH2)2CH3),1.29(s,3H,CH3),1.08(d,J=8.8 Hz,3H,CH3),0.96(s,3H,CH3),0.87(s,3H,CH3),0.75(s,3H,CH3).
3.7 2-肟基-3β,23-O-异亚丙基-121,3-二羟基-28-戊酯的合成(Ⅰ4)
按照制备Ⅰ1的方法,将中间体AA-3(100 mg,0.18 mmol)与溴戊烷(0.2 mL)反应,粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=10∶1,得到化合物Ⅰ4,产率为26.7%.mp 235.7~239.9 ℃.ESI-MS(m/z):646.2(M+H)+.1H-NMR(300 MHz,CDCl3),δ:11.43(s,1H,NOH),4.10(t,J=7.9 Hz,2H,COOCH2CH2CH2CH2CH3),3.89(s,1H,H-3),3.58(d,2H,J=9.6 Hz,H-23),3.44(s,1H,H-12),1.88~1.41(m,4H,COOCH2(CH2)2CH3),1.31(s,3H,CH3),1.13(s,3H,CH3),1.10(d,J=8.8 Hz,3H,CH3),0.98(t,J=7.1 Hz,3H,COOCH2CH2CH2CH2CH3),0.91(s,3H,CH3),0.88(s,3H,CH3).
3.8 N-(2-肟基-3β,23-O-二异亚丙基-12,13-羟基-28酰)-苯胺的合成(Ⅰ5)
将化合物AA-3(100 mg,0.18 mmol)溶解在CH2Cl2(10 mL)中,滴加草酰氯(0.80 mmol)反应24 h,减压蒸除溶剂,加入2 mL环己烷,蒸除环己烷,反复3次,得到化合物AA-5,然后加入CH2Cl2(10 mL)溶解后,再加入少量三乙胺调节酸碱度至微碱性(pH=9~10),在此基础上加入苯胺(0.40 mmol),TLC检测反应终点.减压,蒸除CH2Cl2,加入10 mL水和少量稀盐酸调至酸性(pH=3~4),减压抽滤,用蒸馏水洗滤饼至中性.用硅胶柱色谱进行纯化,洗脱剂为V(石油醚)∶V(乙酸乙酯)=5∶1,得白色固体AA-6-1,产率为46.5%.将CH2Cl2(10 mL)溶解中间产物AA-6-1后,加入间氯过氧苯甲酸(75 mg,0.44 mmol),在室温条件下反应8 h,TLC监测反应终点.用二氯甲烷和饱和食盐水反复萃取3次,然后用饱和NaHCO3洗涤至中性,无水硫酸钠干燥,减压蒸除得到白色固体.白色固体用10 mL氯仿溶解,2 mL浓盐酸搅拌2 h,TLC检测反应终点.用饱和食盐水和CHCl3萃取3次,加入少量NaHCO3溶液洗涤至中性,减压,蒸除溶剂,粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=8∶1,得到化合物Ⅰ5,产率为28.9%.mp 220.3~222.1 ℃.ESI-MS(m/z):651.1(M+H)+.1H-NMR(300 MHz,CDCl3),δ:11.39(s,1H,NOH),7.55~7.09(m,5H,NH(C6H5)),3.77(s,1H,H-3),3.53(d,2H,J=9.6 Hz,H-23),3.44(s,1H,H-12),1.25(s,3H,CH3),1.02(s,3H,CH3),0.92(d,J=8.8 Hz,3H,CH3),0.90(s,3H,CH3),0.89(s,3H,CH3).
3.9 N-(2-肟基-3β,23-O-异亚丙基-12,13-二羟基-28-酰)-间硝基苯胺的合成(Ⅰ6)
按照制备Ⅰ5的方法,由中间体AA-3与间硝基苯胺(0.4 mmol)反应得到粗品,粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=8∶1,得到化合物Ⅰ6,产率为30.2%.mp 215.5~218.6 ℃.ESI-MS(m/z):695.2(M+H)+.1H-NMR(300 MHz,CDCl3),δ:11.41(s,1H,NOH),8.39~7.41(m,4H,NH(C6H4)NO2),3.66(s,1H,H-3),3.53(d,2H,J=9.6 Hz,H-23),3.47(s,1H,H-12),1.04(s,3H,CH3),1.02(s,3H,CH3),0.90(d,J=8.8 Hz,3H,CH3),0.89(s,3H,CH3),0.89(s,3H,CH3).
3.10 N-(2-肟基-3β,23-O-异亚丙基-12,13-二羟基-28-酰)-间氟苯胺的合成(Ⅰ7)
按照制备Ⅰ5的方法,由中间体AA-3与间氟苯胺(0.4 mmol)反应得到粗品.粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=8∶1,得到化合物Ⅰ7,产率为27.1%.mp 213.5~214.9 ℃.1H-NMR(300 MHz,CDCl3),δ:11.43(s,1H,NOH),7.24~6.82(m,4H,NH(C6H4)F),3.90(s,1H,H-3),3.56(d,2H,J=9.6 Hz,H-23),3.46(s,1H,H-12),1.42(s,3H,CH3),1.27(s,3H,CH3),1.08(s,3H,CH3),1.04(s,3H,CH3),0.96(s,J=8.8 Hz,3H,CH3),0.75(d,3H,CH3).
3.11 N-(2-肟基-3β,23-O-异亚丙基-12,13-二羟基-28-酰)-吗啡啉的合成(Ⅰ8)
按照制备Ⅰ5的方法,由中间体AA-3与吗啡啉(0.4 mmol)反应得到粗品.粗品经过柱色谱纯化,V(石油醚)∶V(乙酸乙酯)=8∶1,得到化合物Ⅰ8,产率为29.3%.mp 216.3~218.7 ℃.1H-NMR(300 MHz,CDCl3),δ:11.46(s,1H,NOH),4.52~4.32(m,8H,N(CH2)2(CH2)2O),3.80(s,1H,H-3),3.53(d,2H,J=9.6 Hz,H-23),3.45(s,1H,H-12),1.30(s,3H,CH3),1.27(s,3H,CH3),1.04(s,3H,CH3),0.93(s,3H,CH3),0.87(s,J=8.8 Hz,3H,CH3),0.85(s,3H,CH3).
4 体外活性测试
为考察目标化合物Ⅰ1~Ⅰ8的抗肿瘤活性,选取吉非替尼(Gefitinib)和阿霉素(Adriamycin)作为阳性对照物,人肝癌细胞(HepG2)和人肺癌细胞(A549)为肿瘤细胞,采用MTT法对目标化合物进行活性测试.将对数生长期的肿瘤细胞制成细胞悬液,埋于96孔板中,每孔160 μL,置于37 ℃、含体积分数5%的CO2培养箱中培养24 h.将待测药品配制成不同质量浓度(初始质量浓度为1000 mg/L,稀释后为400 mg/L、40 mg/L、4 mg/L、0.4 mg/L、0.04 mg/L)的药液,各取20 μL加入96孔板内,每个质量浓度3个平行孔.加药完毕后,放入37 ℃、含体积分数5%的CO2培养箱中培养72 h.72 h后,将孔板内培养基吸出,每孔加入MTT溶液(5 g/L)20 μL,37 ℃培养3 h后,弃去上清液,每孔加100 μL的DMSO,用酶标仪在490 nm下测光密度(OD),通过IC50计算软件计算所测化合物的IC50值.不同化合物对HepG2和A549的IC50值如表1所示.
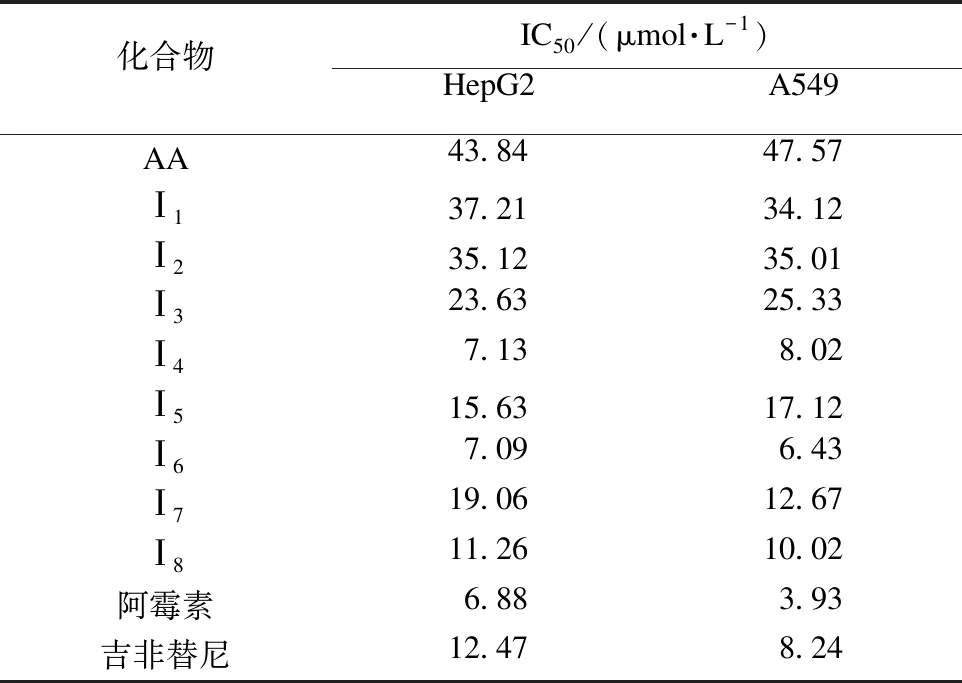
表1 目标化合物对HepG2细胞和A549细胞的活性抑制Table 1 Inhibitory effect of target compound on HepG2 cells and A549 cells
表1结果显示:目标化合物的抗肿瘤活性均显著提高,并优于母体积雪草酸.化合物Ⅰ4、Ⅰ6对HepG2细胞和A549表现出良好的抑制活性.化合物Ⅰ4、Ⅰ6对HepG2细胞的IC50值分别是7.13、7.09 μmol/L,对A549的IC50值分别是8.02、6.43μmol/L,较吉非替尼强,较阿霉素弱.从表1可知通过改造积雪草酸A环上的3个羟基,C-28位羧基以及将C-12位双键打开可以显著提高积雪草酸的生物活性.
5 结 论
笔者设计合成了8种积雪草酸衍生物.体外抗肿瘤活性结果显示:化合物Ⅰ4、Ⅰ6的抗肿瘤活性明显优于其他化合物;在实验范围内,化合物活性Ⅰ4>Ⅰ3>Ⅰ2>Ⅰ1,说明酯链越长,抗肿瘤活性越高;化合物Ⅰ5~Ⅰ8活性明显强于AA,且强于化合物Ⅰ3、Ⅰ2、Ⅰ1,说明C-28位引入胺基时抗肿瘤活性提高.该实验结果对积雪草酸进一步的结构改造和优化具有一定的指导意义,为以后新型抗肿瘤药物的研发提供了基础.
猜你喜欢
佳木斯大学学报(自然科学版)(2020年1期)2020-02-28 05:30:52
食品与机械(2018年5期)2018-07-14 03:15:24
中成药(2018年6期)2018-07-11 03:01:28
中国有色金属学报(2018年2期)2018-03-26 07:58:48
中成药(2017年12期)2018-01-19 02:06:26
中成药(2017年5期)2017-06-13 13:01:12
农产品加工(2017年6期)2017-05-09 18:04:52
橡胶工业(2016年2期)2016-02-23 21:36:51
合成化学(2015年9期)2016-01-17 08:57:14
医学美学美容·中旬刊(2015年2期)2015-10-21 19:58:27
