
HPLC 法测定酚麻美敏口服溶液中对氨基酚的含量
2021-01-29 05:13李滋,刘帅,王卫
天津药学 2020年4期
李 滋,刘 帅,王 卫
(天津市药品检验研究院,天津 300070)
酚麻美敏口服溶液是复方制剂,用于普通感冒、流行性感冒及过敏性疾病等引起的发热、头痛、四肢酸痛、打喷嚏、流鼻涕、咳嗽等症状。该药的主要成分包括对乙酰氨基酚、盐酸伪麻黄碱、氢溴酸右美沙芬及马来酸氯苯那敏。其中,对乙酰氨基酚在制备中由于乙酰化不完全,或在保存过程中发生水解,会生成对氨基酚[1,2]。对氨基酚毒性较大,可能会作用于肾脏血液等而引发相关的疾病或损伤,且具有一定的基因毒性[3,4]。《中国药典》2015 年版二部[5]的对乙酰氨基酚及其单方制剂项下均收录了对氨基酚检查项。而酚麻美敏口服溶液的现行质量标准[国家食品药品监督管理总局国家药品标准WS-106(X-093)-2002-2015Z]中并没有对对氨基酚的含量进行控制。本试验采用高效液相色谱法测定酚麻美敏口服溶液中对氨基酚的含量,专属性强,灵敏度高,具有较好的重复性和准确性。
1 仪器与试药
HP-1100 高效液相色谱仪,华谱S6000 高效液相色谱仪,XSE 205 分析天平(METTLER TOLEDO),GL Sciences Inertsil C8柱(250 mm×4.6 mm,5 μm),Agilent ZORBAX Eclipse Plus-C8柱(250 mm×4.6 mm,5 μm);对氨基酚对照品(批号100802-201604),对乙酰氨基酚对照品(批号100018-201610),盐酸伪麻黄碱对照品(批号171237-201510),氢溴酸右美沙芬对照品(批号100201-201201),马来酸氯苯那敏对照品(批号100047-201507),均来自中国食品药品检定研究院;酚麻美敏口服溶液样品(每毫升含对乙酰氨基酚32 mg、盐酸伪麻黄碱3 mg、氢溴酸右美沙芬1 mg 和马来酸氯苯那敏0.2 mg),来自于青岛正大海尔制药有限公司,批号分别为1901031、1901041、1901051;甲醇为色谱 纯(Merck 公司),其余试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱条件 采用HP-1100 高效液相色谱仪,GL Sciences Inertsil C8色谱柱(250 mm×4.6 mm,5 μm),流动相为磷酸盐缓冲液(取磷酸氢二钠8.95 g,磷酸二氢钠3.9 g,加水溶解至1 000 ml,加10%四丁基氢氧化铵溶液12 ml)-甲醇(90∶10),流速:1.0 ml/min,柱温:40 ℃,检测波长:245 nm,进样20 μl。
2.2 溶液制备
2.2.1 对照品贮备液的制备 临用新制。取对氨基酚对照品约16 mg,置50 ml 量瓶中,加溶剂[甲醇-水(4 ∶6)]溶解并稀释至刻度,摇匀。
2.2.2 对照品溶液的制备 临用新制。分别取对氨基酚对照品与对乙酰氨基酚对照品16 mg,置同一100 ml量瓶中,加溶剂溶解并稀释至刻度,摇匀;精密量取2 ml,置50 ml 量瓶中,加溶剂稀释至刻度,摇匀。
2.2.3 供试品溶液的制备 临用新制。用内容量移液管精密量取本品5 ml,置25 ml 量瓶中,用溶剂分次洗涤移液管内壁,洗液并入量瓶中,用溶剂稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
2.2.4 空白辅料溶液的制备 按处方比例,制备不含四种主成分的辅料溶液。按“2.2.3”项下方法制备空白辅料溶液。
2.2.5 阴性对照溶液 按处方比例取盐酸伪麻黄碱、氢溴酸右美沙芬及马来酸氯苯那敏制备阴性样品,并按照“2.2.3”项下方法制备阴性对照溶液。
2.3 专属性考查 按照“2.1”项下的色谱条件,精密量取对照品溶液、供试品溶液、空白辅料溶液及阴性对照溶液各20 μl,注入液相色谱仪,结果表明对氨基酚与对乙酰氨基酚的分离度为21.8,理论板数按对乙酰氨基酚计为13 667,空白辅料溶液及其他成分对对氨基酚的测定无干扰,见图1。
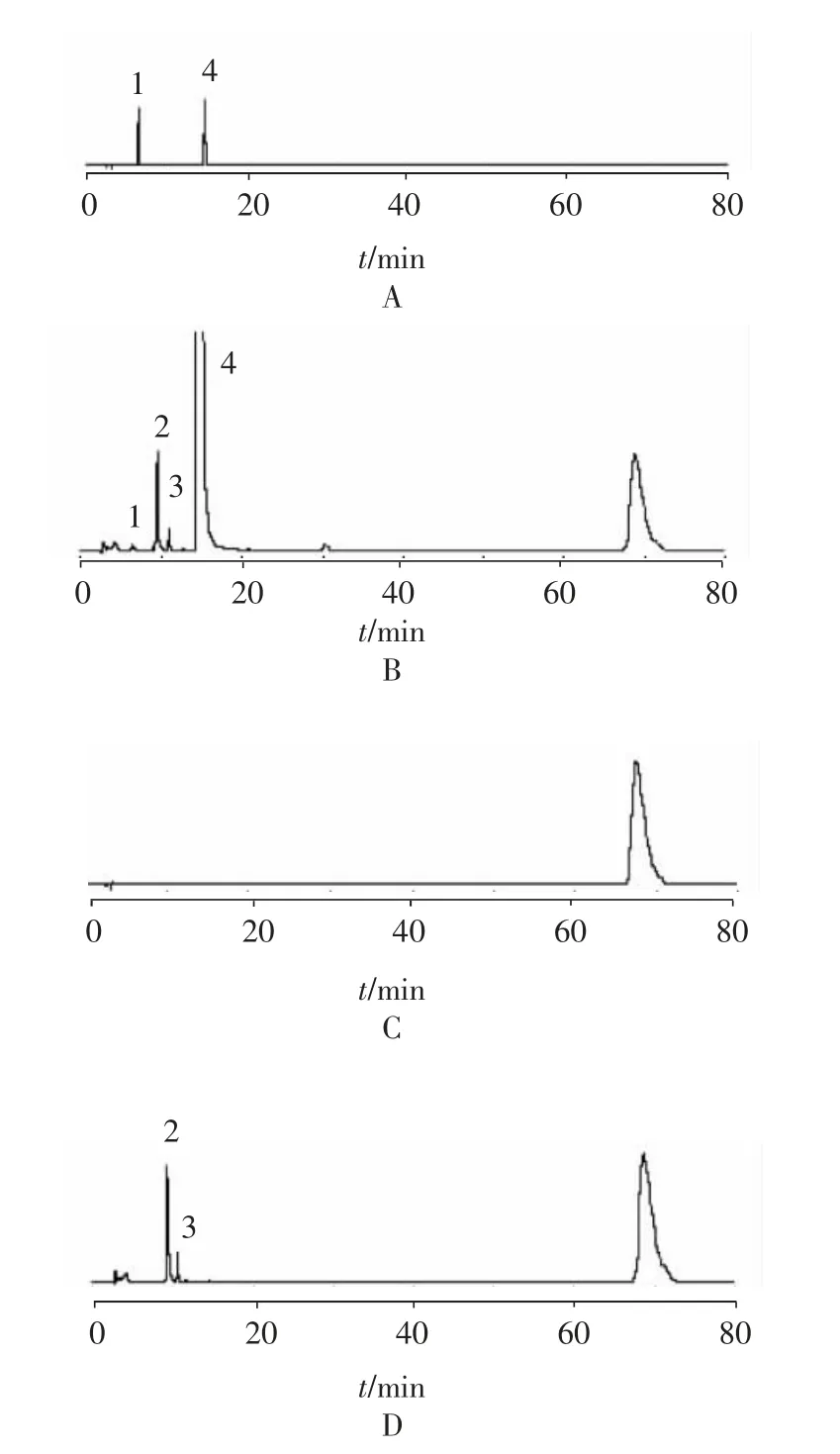
图1 对照品(A)供试品(B)空白辅料(C)阴性对照(D)HPLC 色谱图
2.4 破坏试验 精密量取供试品5 ml,置25 ml 量瓶中,进行不同条件下的降解试验。①酸破坏:加1 mol/L盐酸溶液2 ml,摇匀,放置30 min,用1 mol/L 氢氧化钠溶液调至pH 值为中性,加溶剂稀释至刻度,摇匀,滤过。②碱破坏:加1 mol/L 氢氧化钠溶液2 ml,摇匀,放置30 min,用1 mol/L 盐酸溶液调至pH 值为中性,加溶剂稀释至刻度,摇匀,滤过。③氧化破坏:加10%过氧化氢溶液2 ml,摇匀,放置30 min,加溶剂稀释至刻度,摇匀,滤过。④高温破坏:加溶剂适量,摇匀,80 ℃水浴中加热1 h,放冷,加溶剂至刻度,摇匀,滤过。⑤光照破坏:加溶剂稀释至刻度,直接光照5 d 后摇匀,滤过。分别取上述5 种续滤液,按“2.1”项色谱条件测定。结果见图2。结果表明:上述破坏条件下产生的其他杂质峰与对氨基酚峰可以达到基线分离,不影响对氨基酚的测定,说明拟定的色谱条件专属性较好。
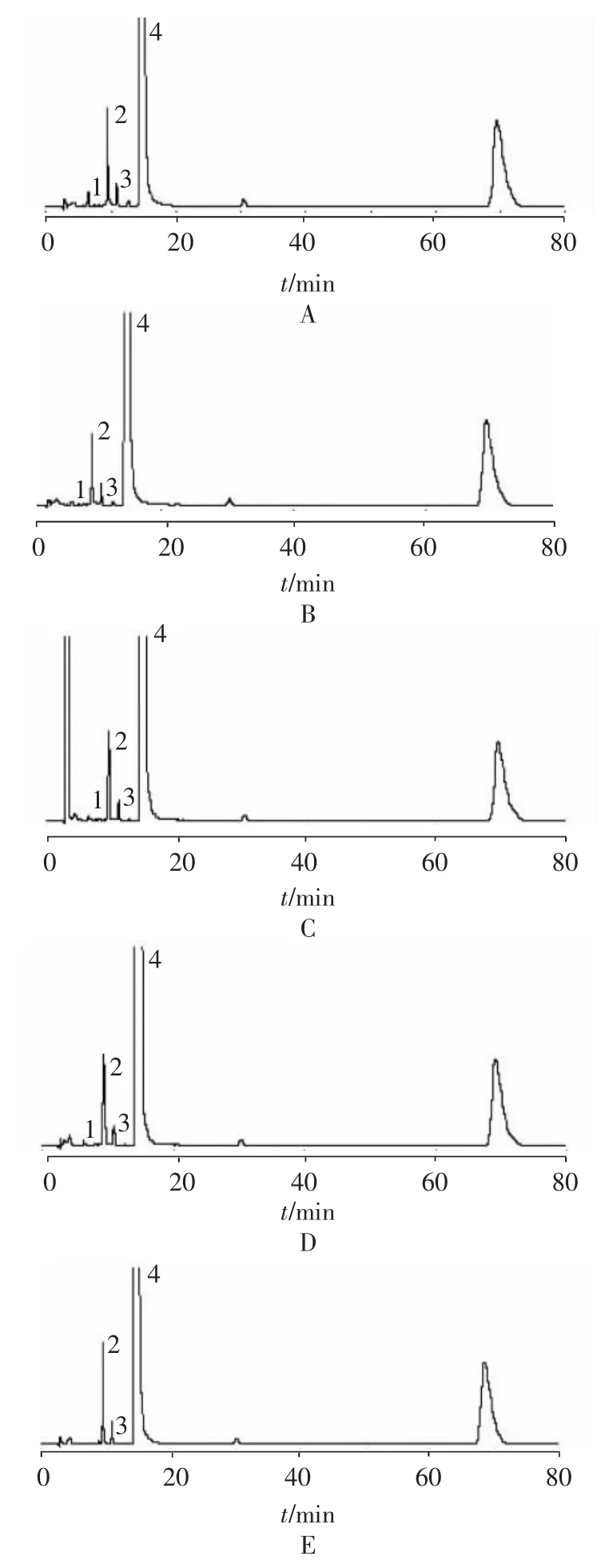
图2 酸破坏(A)碱破坏(B)氧化破坏(C)高温破坏(D)光照破坏(E)HPLC 色谱图
2.5 线性关系考查 精密量取对照品贮备液适量,加溶剂稀释制成每1 ml 中含对氨基酚0.64、1.6、3.2、6.4和12.8 μg 的系列溶液,按“2.1”项下色谱条件进样,记录色谱图。以浓度为横坐标(C),以峰面积为纵坐标(A),进行线性回归。结果表明对氨基酚在0.64~12.8 μg/ml浓度范围内与峰面积呈良好线性关系,线性方程为A=35.521 C-0.8686(r=0.9999,n=5)。
2.6 重现性试验 取同一批样品(批号1901051)依“2.2.3”项下方法制备6 份供试品溶液,依“2.1”项下色谱条件进样测定。结果对氨基酚的平均含量为0.016%,RSD 为1.2%,表明该方法重现性良好。
2.7 稳定性试验 取对照品溶液和供试品溶液(批号1901051)在室温条件下分别放置0、2、4、6、8、10、12 和24 h,依“2.1”项下的色谱条件进样。结果在8 h 内,对照品溶液和供试品溶液中对氨基酚峰面积的RSD 分别为1.6%和0.4%,说明对照品溶液及供试品溶液在8 h 内基本稳定。
2.8 检出限试验 精密量取对照品溶液适量,采用逐步稀释法测定,按照S/N=3 计算,对氨基酚的检出限为0.32 μg/ml。
2.9 回收率试验 精密量取对照品贮备液5 ml,置50 ml 量瓶中,加溶剂稀释至刻度,摇匀,作为浓对照品溶液。分别精密量取供试品(批号1901031,未检出对氨基酚)5 ml 置9 个25 ml 量瓶中,分别精密加入浓对照品溶液4、5 和6 ml 各3 份,用溶剂稀释至刻度,摇匀,滤过,取续滤液依“2.1”项下的色谱条件进样,记录色谱图,计算平均回收率。结果平均回收率为99.7%,RSD 为0.8%,见表1。
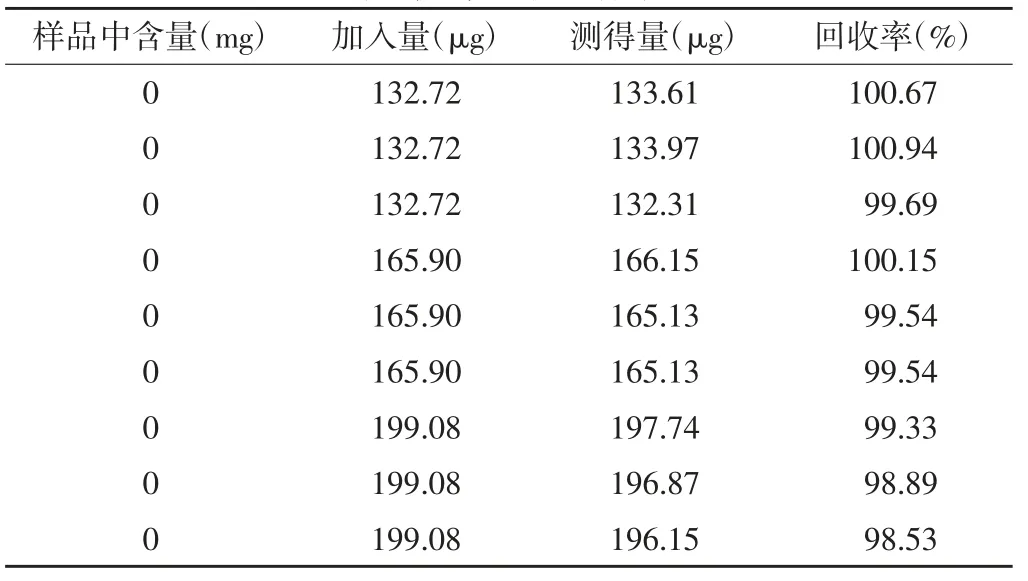
表1 回收率试验结果(n=9)
2.10 耐用性试验 采用不同的厂家的仪器和色谱柱进行耐用性考查。条件1:HP-1100 高效液相色谱仪,GL Sciences Inertsil C8色谱柱(250 mm×4.6 mm,5 μm);条件2:华谱S6000 液相色谱仪,Agilent ZORBAX Eclipse Plus-C8色谱柱(250 mm×4.6 mm,5 μm)。试验结果见表2,结果基本一致,表明该方法的耐用性良好。

表2 耐用性试验结果
2.11 样品测定 照“2.2.1”项下方法制备供试品溶液,照“2.1”项下色谱条件进行测定,计算供试品中对氨基酚的含量,结果见表3。

表3 含量测定结果
3 讨论
3.1 流动相的考查 文献资料中采用了不同种类的流动相[6-8],为了达到更好的分离效果,最终采用磷酸盐缓冲液(取磷酸氢二钠8.95 g,磷酸二氢钠3.9 g,加水溶解至1 000 ml,加10%四丁基氢氧化铵溶液12 ml)-甲醇(90 ∶10)为流动相,各峰能够达到基线分离,满足对氨基酚含量测定的要求。
3.2 供试品溶液的制备 《中国药典》2015 年版二部对乙酰氨基酚质量标准对氨基酚检查项下,供试品溶液的浓度为20 mg/ml。本品若按20 mg/ml 浓度配制供试品溶液,则需精密量取本品15 ml 置25 ml 量瓶中并进行定容操作。由于本品为黏稠液体,需分次清洗移液管并将洗液一并转移至量瓶中,会导致溶液总量易超出量瓶的刻度范围。经试验,拟定方法为“用内容量移液管精密量取本品5 ml,置25 ml 量瓶中,用溶剂分次洗涤移液管内壁,洗液并入量瓶中,用溶剂稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液”,其浓度为6.4 mg/ml,相应对照品溶液的浓度也调整为6.4 μg/ml。
3.3 限量的确定 《中国药典》2015 年版的对乙酰氨基酚制剂中对氨基酚检查项的限度均为0.1%,经参考该限度并结合实际的试验结果,将对氨基酚的限度拟定为0.1%。
采用HPLC 法测定酚麻美敏口服溶液中对氨基酚含量,灵敏度高,精密度好,具有较好的专属性及准确性,适用于酚麻美敏口服溶液的质量控制。
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
农药学学报(2022年3期)2022-06-14
保健与生活(2022年12期)2022-06-09
节能与环保(2022年3期)2022-04-26
安徽农学通报(2022年6期)2022-04-07
药品评价(2021年17期)2021-11-06
保健与生活(2020年4期)2020-03-02
家庭科学·新健康(2018年11期)2018-11-16
发明与创新·中学生(2017年1期)2017-01-20
家庭用药(2016年8期)2016-05-14
